false
FY
0001106838
0001106838
2022-10-01
2023-09-30
0001106838
2023-03-31
0001106838
2023-12-11
0001106838
2023-09-30
0001106838
2022-09-30
0001106838
us-gaap:RelatedPartyMember
2023-09-30
0001106838
us-gaap:RelatedPartyMember
2022-09-30
0001106838
2021-10-01
2022-09-30
0001106838
us-gaap:PreferredStockMember
2021-09-30
0001106838
us-gaap:CommonStockMember
2021-09-30
0001106838
us-gaap:AdditionalPaidInCapitalMember
2021-09-30
0001106838
us-gaap:RetainedEarningsMember
2021-09-30
0001106838
2021-09-30
0001106838
us-gaap:PreferredStockMember
2022-09-30
0001106838
us-gaap:CommonStockMember
2022-09-30
0001106838
us-gaap:AdditionalPaidInCapitalMember
2022-09-30
0001106838
us-gaap:RetainedEarningsMember
2022-09-30
0001106838
us-gaap:PreferredStockMember
2021-10-01
2022-09-30
0001106838
us-gaap:CommonStockMember
2021-10-01
2022-09-30
0001106838
us-gaap:AdditionalPaidInCapitalMember
2021-10-01
2022-09-30
0001106838
us-gaap:RetainedEarningsMember
2021-10-01
2022-09-30
0001106838
us-gaap:PreferredStockMember
2022-10-01
2023-09-30
0001106838
us-gaap:CommonStockMember
2022-10-01
2023-09-30
0001106838
us-gaap:AdditionalPaidInCapitalMember
2022-10-01
2023-09-30
0001106838
us-gaap:RetainedEarningsMember
2022-10-01
2023-09-30
0001106838
us-gaap:PreferredStockMember
2023-09-30
0001106838
us-gaap:CommonStockMember
2023-09-30
0001106838
us-gaap:AdditionalPaidInCapitalMember
2023-09-30
0001106838
us-gaap:RetainedEarningsMember
2023-09-30
0001106838
SONN:OctoberOfferingMember
SONN:FebruaryTwoThousandTwentyFourMember
2022-10-01
2023-09-30
0001106838
2023-01-01
2023-09-30
0001106838
SONN:OctoberOfferingMember
SONN:UnderwritersMember
2023-10-26
2023-10-26
0001106838
SONN:OctoberOfferingMember
us-gaap:CommonStockMember
SONN:UnderwritersMember
2023-10-26
2023-10-26
0001106838
SONN:OctoberOfferingMember
SONN:PrefundedWarrantMember
SONN:UnderwritersMember
2023-10-26
0001106838
SONN:OctoberOfferingMember
SONN:CommonWarrantMember
SONN:UnderwritersMember
2023-10-26
0001106838
SONN:OctoberOfferingMember
us-gaap:CommonStockMember
SONN:UnderwritersMember
2023-10-26
0001106838
SONN:OctoberOfferingMember
SONN:UnderwritersMember
2023-10-26
0001106838
SONN:OctoberOfferingMember
2023-09-30
0001106838
SONN:SalesAgreementMember
2022-09-30
0001106838
SONN:ResearchAndDevelopmentTaxIncentiveProgramMember
2022-10-01
2023-09-30
0001106838
SONN:ResearchAndDevelopmentTaxIncentiveProgramMember
2021-10-01
2022-09-30
0001106838
2023-01-01
2023-01-31
0001106838
2023-04-01
2023-04-30
0001106838
2023-08-31
2023-08-31
0001106838
SONN:CommonStockWarrantsAugustTwentyTwentyOneMember
2022-10-01
2023-09-30
0001106838
SONN:CommonStockWarrantsAugustTwentyTwentyOneMember
2021-10-01
2022-09-30
0001106838
SONN:UnderwriterWarrantsAugustTwentyTwentyOneMember
2022-10-01
2023-09-30
0001106838
SONN:UnderwriterWarrantsAugustTwentyTwentyOneMember
2021-10-01
2022-09-30
0001106838
SONN:PrivateWarrantsMember
2022-10-01
2023-09-30
0001106838
SONN:PrivateWarrantsMember
2021-10-01
2022-09-30
0001106838
SONN:ChanticleerWarrantsMember
2022-10-01
2023-09-30
0001106838
SONN:ChanticleerWarrantsMember
2021-10-01
2022-09-30
0001106838
SONN:SeriesCWarrantsMember
2022-10-01
2023-09-30
0001106838
SONN:SeriesCWarrantsMember
2021-10-01
2022-09-30
0001106838
SONN:SeriesThreeWarrantsMember
2022-10-01
2023-09-30
0001106838
SONN:SeriesThreeWarrantsMember
2021-10-01
2022-09-30
0001106838
SONN:UnvestedRestrictedStockUnitsAndAwardsMember
2022-10-01
2023-09-30
0001106838
SONN:UnvestedRestrictedStockUnitsAndAwardsMember
2021-10-01
2022-09-30
0001106838
SONN:CommonStockWarrantsFebruaryTwentyTwentyThreeMember
2022-10-01
2023-09-30
0001106838
SONN:CommonStockWarrantsFebruaryTwentyTwentyThreeMember
2021-10-01
2022-09-30
0001106838
SONN:UnderwriterWarrantsFebruaryTwentyTwentyThreeMember
2022-10-01
2023-09-30
0001106838
SONN:UnderwriterWarrantsFebruaryTwentyTwentyThreeMember
2021-10-01
2022-09-30
0001106838
SONN:CommonStockPrivatePlacementWarrantsJuneTwentyTwentyThreeMember
2022-10-01
2023-09-30
0001106838
SONN:CommonStockPrivatePlacementWarrantsJuneTwentyTwentyThreeMember
2021-10-01
2022-09-30
0001106838
SONN:PAWarrantsJuneTwentyTwentyThreeMember
2022-10-01
2023-09-30
0001106838
SONN:PAWarrantsJuneTwentyTwentyThreeMember
2021-10-01
2022-09-30
0001106838
SONN:DiscoveryCollaborationAgreementsMember
SONN:XOMAMember
2012-07-31
0001106838
SONN:DiscoveryCollaborationAgreementsMember
SONN:XOMAMember
us-gaap:InProcessResearchAndDevelopmentMember
2022-04-01
2022-04-30
0001106838
SONN:DiscoveryCollaborationAgreementsMember
SONN:XOMAMember
us-gaap:InProcessResearchAndDevelopmentMember
2022-10-01
2023-09-30
0001106838
SONN:CellcaAgreementMember
2022-10-01
2023-09-30
0001106838
SONN:CellcaAgreementMember
srt:MaximumMember
2019-01-31
0001106838
SONN:TheCellcaAgreementMember
srt:MinimumMember
2023-09-30
0001106838
SONN:TheCellcaAgreementMember
srt:MaximumMember
2023-09-30
0001106838
SONN:TheCellcaAgreementMember
2023-09-30
0001106838
SONN:TheCellcaAgreementMember
us-gaap:InProcessResearchAndDevelopmentMember
2022-04-30
0001106838
SONN:TheCellcaAgreementMember
us-gaap:InProcessResearchAndDevelopmentMember
2022-10-01
2023-09-30
0001106838
SONN:TheBrinkAgreementMember
us-gaap:InProcessResearchAndDevelopmentMember
2023-09-30
0001106838
SONN:TheBrinkAgreementMember
2023-04-30
0001106838
SONN:TheBrinkAgreementMember
2023-09-30
0001106838
SONN:TheBrinkAgreementMember
us-gaap:InProcessResearchAndDevelopmentMember
2022-10-01
2023-09-30
0001106838
SONN:TheBrinkAgreementMember
us-gaap:InProcessResearchAndDevelopmentMember
2021-10-01
2022-09-30
0001106838
SONN:InvivoGenAgreementMember
2023-09-30
0001106838
SONN:InvivoGenAgreementMember
us-gaap:InProcessResearchAndDevelopmentMember
2022-10-01
2023-09-30
0001106838
SONN:InvivoGenAgreementMember
2022-10-01
2023-09-30
0001106838
SONN:ProteoNicAgreementMember
us-gaap:InProcessResearchAndDevelopmentMember
2022-03-01
2022-03-31
0001106838
SONN:ProteoNicAgreementMember
2023-09-30
0001106838
SONN:ProteoNicAgreementMember
us-gaap:InProcessResearchAndDevelopmentMember
2022-10-01
2023-09-30
0001106838
SONN:ProteoNicAgreementMember
2022-10-01
2023-09-30
0001106838
SONN:NavigoAgreementMember
SONN:NavigoProteinsGmbHMember
us-gaap:TechnologyServiceMember
2021-10-01
2022-09-30
0001106838
SONN:NavigoAgreementMember
SONN:NavigoProteinsGmbHMember
us-gaap:TechnologyServiceMember
2023-03-31
0001106838
SONN:NavigoAgreementMember
SONN:NavigoProteinsGmbHMember
us-gaap:TechnologyServiceMember
2023-09-30
0001106838
SONN:LicenseAgreementMember
2020-08-01
2020-08-31
0001106838
SONN:NewLifeAgreementMember
2021-06-01
2021-06-30
0001106838
SONN:NewLifeAgreementMember
2023-09-30
0001106838
SONN:NewLifeAgreementMember
srt:MinimumMember
2023-09-30
0001106838
SONN:NewLifeAgreementMember
srt:MaximumMember
2023-09-30
0001106838
SONN:SalesAgreementMember
SONN:BTIGLLCMember
2022-08-15
2022-08-15
0001106838
SONN:SalesAgreementMember
2022-08-15
2022-08-15
0001106838
SONN:SalesAgreementMember
SONN:BTIGLLCMember
2022-10-01
2023-09-30
0001106838
SONN:SalesAgreementMember
2023-02-10
2023-02-10
0001106838
SONN:SalesAgreementMember
us-gaap:CommonStockMember
SONN:InvestorsMember
2023-02-10
2023-02-10
0001106838
SONN:SalesAgreementMember
SONN:PrefundedWarrantMember
SONN:InvestorsMember
2023-02-10
0001106838
SONN:SalesAgreementMember
SONN:CommonWarrantMember
SONN:InvestorsMember
2023-02-10
0001106838
SONN:SalesAgreementMember
SONN:UnderwriterWarrantsMember
2023-02-10
0001106838
SONN:ChardanCapitalMarketsLLCMember
2023-06-01
2023-06-30
0001106838
SONN:ChardanCapitalMarketsLLCMember
2023-06-30
2023-06-30
0001106838
SONN:SalesAgreementMember
SONN:PrefundedWarrantMember
SONN:ChardanCapitalMarketsLLCMember
2023-06-30
0001106838
SONN:ChardanCapitalMarketsLLCMember
2023-06-30
0001106838
SONN:SalesAgreementMember
SONN:CommonWarrantMember
SONN:ChardanCapitalMarketsLLCMember
srt:ScenarioForecastMember
2023-12-30
0001106838
SONN:SalesAgreementMember
SONN:CommonWarrantMember
SONN:ChardanCapitalMarketsLLCMember
2023-09-30
0001106838
SONN:SalesAgreementMember
SONN:UnderwriterWarrantsMember
2023-09-30
0001106838
SONN:SalesAgreementMember
SONN:UnderwriterWarrantsMember
srt:ScenarioForecastMember
2023-12-30
0001106838
SONN:SecuritiesPurchaseAgreementMember
SONN:AccreditedInvestorsMember
SONN:SeriesThreeConvertiblePreferredStockMember
2022-08-01
2022-08-31
0001106838
SONN:SecuritiesPurchaseAgreementMember
SONN:AccreditedInvestorsMember
SONN:SeriesThreeConvertiblePreferredStockMember
2022-08-31
0001106838
SONN:SecuritiesPurchaseAgreementMember
SONN:AccreditedInvestorsMember
SONN:SeriesFourConvertiblePreferredStockMember
2022-08-01
2022-08-31
0001106838
SONN:SecuritiesPurchaseAgreementMember
SONN:AccreditedInvestorsMember
SONN:SeriesFourConvertiblePreferredStockMember
2022-08-31
0001106838
SONN:SecuritiesPurchaseAgreementMember
SONN:AccreditedInvestorsMember
SONN:SeriesThreeWarrantsMember
2022-08-31
0001106838
SONN:SecuritiesPurchaseAgreementMember
SONN:AccreditedInvestorsMember
SONN:SeriesThreeWarrantsMember
2022-08-01
2022-08-31
0001106838
SONN:SecuritiesPurchaseAgreementMember
SONN:AccreditedInvestorsMember
SONN:SeriesThreeWarrantsMember
us-gaap:PrivatePlacementMember
2022-08-01
2022-08-31
0001106838
SONN:SecuritiesPurchaseAgreementMember
SONN:AccreditedInvestorsMember
2022-09-15
2022-09-15
0001106838
SONN:SecuritiesPurchaseAgreementMember
SONN:AccreditedInvestorsMember
SONN:SeriesThreeConvertiblePreferredStockMember
2022-09-30
2022-09-30
0001106838
SONN:SecuritiesPurchaseAgreementMember
SONN:AccreditedInvestorsMember
SONN:SeriesFourConvertiblePreferredStockMember
2022-09-30
2022-09-30
0001106838
SONN:TwentyTwentyTwoSalesAgeementMember
SONN:BTIGMember
us-gaap:CommonStockMember
2021-10-01
2022-09-30
0001106838
SONN:CommonStockWarrantsMember
2022-10-01
2023-09-30
0001106838
SONN:PrefundedWarrantMember
2022-10-01
2023-09-30
0001106838
SONN:CommonStockWarrantsAugustTwentyTwentyOneMember
2023-09-30
0001106838
SONN:UnderwriterWarrantsAugustTwentyTwentyOneMember
2023-09-30
0001106838
SONN:ChanticleerWarrantsMember
2023-09-30
0001106838
SONN:ChanticleerWarrantsMember
srt:MinimumMember
2023-09-30
0001106838
SONN:ChanticleerWarrantsMember
srt:MaximumMember
2023-09-30
0001106838
SONN:SeriesCWarrantsMember
2023-09-30
0001106838
SONN:Series3WarrantsMember
2023-09-30
0001106838
SONN:CommonStockWarrantsFebruaryTwentyTwentyThreeMember
2023-09-30
0001106838
SONN:UnderwriterWarrantFebruaryTwentyTwentyThreeMember
2023-09-30
0001106838
SONN:CommonStockPrivatePlacementWarrantsJuneTwentyTwentyThreeMember
2023-09-30
0001106838
SONN:PAWarrantsJuneTwentyTwentyThreeMember
2023-09-30
0001106838
us-gaap:WarrantMember
2023-09-30
0001106838
SONN:TwentyTwentyOmnibusEquityIncentivePlanMember
us-gaap:CommonStockMember
2023-01-01
0001106838
SONN:TwentyTwentyOmnibusEquityIncentivePlanMember
us-gaap:CommonStockMember
2023-09-30
0001106838
us-gaap:RestrictedStockUnitsRSUMember
2020-07-01
2020-07-31
0001106838
us-gaap:RestrictedStockUnitsRSUMember
2021-03-01
2021-03-31
0001106838
us-gaap:RestrictedStockUnitsRSUMember
2021-12-01
2021-12-31
0001106838
us-gaap:RestrictedStockUnitsRSUMember
2022-12-01
2022-12-31
0001106838
us-gaap:RestrictedStockUnitsRSUMember
2023-01-01
2023-01-31
0001106838
us-gaap:RestrictedStockUnitsRSUMember
2023-09-30
0001106838
us-gaap:RestrictedStockUnitsRSUMember
srt:MaximumMember
2022-10-01
2023-09-30
0001106838
SONN:RestrictedStockAwardsMember
2023-09-30
0001106838
SONN:RestrictedStockAwardsMember
srt:MaximumMember
2022-10-01
2023-09-30
0001106838
us-gaap:RestrictedStockUnitsRSUMember
us-gaap:ResearchAndDevelopmentExpenseMember
2022-10-01
2023-09-30
0001106838
us-gaap:RestrictedStockUnitsRSUMember
us-gaap:ResearchAndDevelopmentExpenseMember
2021-10-01
2022-09-30
0001106838
us-gaap:RestrictedStockUnitsRSUMember
us-gaap:GeneralAndAdministrativeExpenseMember
2022-10-01
2023-09-30
0001106838
us-gaap:RestrictedStockUnitsRSUMember
us-gaap:GeneralAndAdministrativeExpenseMember
2021-10-01
2022-09-30
0001106838
us-gaap:RestrictedStockUnitsRSUMember
2021-09-30
0001106838
us-gaap:RestrictedStockUnitsRSUMember
2021-10-01
2022-09-30
0001106838
us-gaap:RestrictedStockUnitsRSUMember
2022-09-30
0001106838
us-gaap:RestrictedStockUnitsRSUMember
2022-10-01
2023-09-30
0001106838
SONN:RestrictedStockAwardsMember
2022-09-30
0001106838
SONN:RestrictedStockAwardsMember
2022-10-01
2023-09-30
0001106838
us-gaap:DomesticCountryMember
2023-09-30
0001106838
us-gaap:StateAndLocalJurisdictionMember
2023-09-30
0001106838
us-gaap:ForeignCountryMember
2023-09-30
0001106838
us-gaap:ForeignCountryMember
2022-10-01
2023-09-30
0001106838
srt:MinimumMember
2022-08-31
0001106838
2022-08-31
0001106838
us-gaap:SubsequentEventMember
2023-12-01
2023-12-31
iso4217:USD
xbrli:shares
iso4217:USD
xbrli:shares
xbrli:pure
iso4217:EUR
UNITED
STATES
SECURITIES
AND EXCHANGE COMMISSION
WASHINGTON,
D.C. 20549
FORM
10-K
(Mark
One)
| ☒ |
ANNUAL
REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For
the fiscal year ended September 30, 2023
Or
| ☐ |
TRANSITION
REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For
the transition period from to
Commission
file number: 001-35570
SONNET
BIOTHERAPEUTICS HOLDINGS, INC.
(Exact
name of registrant as specified in its charter)
| Delaware |
|
20-2932652 |
(State
or other jurisdiction of
incorporation
or organization) |
|
(I.R.S.
Employer
Identification
No.) |
| 100
Overlook Center, Suite 102 |
|
|
| Princeton,
NJ |
|
08540 |
| (Address of principal
executive offices) |
|
(Zip Code) |
Registrant’s
telephone number, including area code: (609) 375-2227
Securities
registered pursuant to Section 12(b) of the Act:
| Title
of each class |
|
Trading
Symbol(s) |
|
Name
of each exchange on which registered |
| Common Stock, $0.0001
par value per share |
|
SONN |
|
The Nasdaq Stock Market
LLC |
Securities
registered pursuant to Section 12(g) of the Act: None
Indicate
by check if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes
☐ No ☒
Indicate
by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate
by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange
Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2)
has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate
by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule
405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant
was required to submit such files). Yes ☒ No ☐
Indicate
by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting
company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,”
“smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer |
☐ |
Accelerated filer |
☐ |
| Non-accelerated filer |
☒ |
Smaller reporting company |
☒ |
| |
|
Emerging growth company |
☐ |
If
an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying
with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate
by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness
of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered
public accounting firm that prepared or issued its audit report. ☐
If
securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant
included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate
by check mark if the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☒
The
aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant was $7.3 million on March
31, 2023, the last business day of the registrant’s most recently completed second fiscal quarter, based on the closing price of
$7.92 on that date.
Indicate
the number of shares outstanding of each of the registrant’s classes of common equity, as of December 11, 2023:
| Class |
|
Number
of Shares |
| Common Stock, $.0001
par value |
|
3,069,516 |
Documents
incorporated by reference
None.
TABLE
OF CONTENTS
SPECIAL
NOTE REGARDING FORWARD-LOOKING STATEMENTS
This
report contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Forward-looking
statements include all statements that are not historical facts. In some cases, you can identify forward-looking statements by terms
such as “may,” “will,” “should,” “could,” “would,” “expects,”
“plans,” “anticipates,” “believes,” “estimates,” “projects,” “predicts,”
“potential,” or the negative of those terms, and similar expressions and comparable terminology intended to identify forward-looking
statements. These statements reflect the Company’s current views with respect to future events and are based on assumptions and
subject to risks and uncertainties including those set forth below and under Part I, Item 1A, “Risk Factors” in this annual
report on Form 10-K. Given these uncertainties, you should not place undue reliance on these forward-looking statements. These forward-looking
statements represent the Company’s estimates and assumptions only as of the date of this annual report on Form 10-K and, except
as required by law, the Company undertakes no obligation to update or review publicly any forward-looking statements, whether as a result
of new information, future events or otherwise after the date of this annual report on Form 10-K. You should read this annual report
on Form 10-K and the documents referenced in this annual report on Form 10-K and filed as exhibits completely and with the understanding
that the Company’s actual future results may be materially different from what the Company expects. The Company qualifies all of
its forward-looking statements by these cautionary statements. Such statements may include, but are not limited to, statements concerning
the following:
●
our lack of operating history and history of operating losses;
●
our need for significant additional capital and our ability to satisfy our capital needs;
●
our ability to complete required clinical trials of our products and obtain approval from the FDA or other regulatory agents in different
jurisdictions;
●
our ability to maintain or protect the validity of our patents and other intellectual property;
●
our ability to retain key executive members;
●
our ability to internally develop new inventions and intellectual property;
●
interpretations of current laws and the passages of future laws;
●
acceptance of our business model by investors;
●
the accuracy of our estimates regarding expenses and capital requirements; and
●
our ability to adequately support growth.
PART
I
Item
1. Business.
Overview
Sonnet
BioTherapeutics Holdings, Inc., is a clinical stage, oncology-focused biotechnology company with a proprietary platform for innovating
biologic medicines of single- or bifunctional action. Known as FHAB™ (Fully Human Albumin Binding), the technology utilizes
a fully human single chain antibody fragment that binds to and “hitch-hikes” on human serum albumin (HSA) for transport to
target tissues. We designed the FHAB construct to improve drug accumulation in tumors, as well as to extend the duration of
activity in the body. FHAB development candidates are produced in a mammalian cell culture, which enables glycosylation and
a biological structure similar to the natural cytokines in vivo. We believe our FHAB technology, for which we received
a U.S. patent in June 2021, is a distinguishing feature of our biopharmaceutical platform that is well suited for future drug development
across a range of human disease areas, including oncology, autoimmune, pathogenic, inflammatory, and hematological conditions.
Our
current internal pipeline development activities are focused on cytokines, a class of cell signaling peptides that, among other important
functions, serve as potent immunomodulatory agents. Working both independently and synergistically, specific cytokines have shown the
ability to modulate the activation and maturation of immune cells that fight cancer and pathogens. However, cytokines on their own do
not preferentially accumulate in specific tissues and are quickly eliminated from the body. The conventional approach to achieving a
treatment effect with cytokine therapy typically requires the administration of high and frequent doses. This can result in a reduced
treatment effect accompanied by the potential for systemic toxicity, which poses challenges to the therapeutic application of this class
of drugs.
Sonnet
has built an efficient R&D platform that includes a network of outsourced vendors to help remediate expenses and improve execution
timelines. Most of the vendors are strategic collaborators that offer the company a preferred status with negotiated costs. The major
advantages of this approach include optimized direct investment into projects with expenses that can be rapidly scaled up or down depending
on the number of projects. The cost advantages of the Sonnet platform start at the vendor network selection process, with CMC being one
of the most expensive components of the initial drug development step. Sonnet has chosen a strategic CMC collaborator in India and has
negotiated the cost to be significantly less than the expense incurred from a similar US- or Europe-based vendor. Sonnet is conducting
two of the company’s three ongoing clinical trials in Australia, which carries a substantial cost reduction relative to US trials
via the Australian government’s R&D tax credit program. Sonnet is also coordinating the Indian and Australian execution with
top R&D vendors from the US, England, Germany, and Switzerland, with the objective of directing the bulk of the company’s operating
expense infrastructure towards its drug development pipeline.
Pipeline
We
have a pipeline of therapeutic compounds focused primarily on oncology indications of high unmet medical need.
| |
● |
Our
lead proprietary asset, SON-1010, is a fully human single-chain version of Interleukin 12 (IL-12), covalently linked to the FHAB
construct, for which we are pursuing clinical development in solid tumors. Sonnet has completed a non-human primate (NHP) toxicity
study, conducted under current Good Laboratory Practices (cGLP), and has successfully manufactured both liquid and lyophilized forms
of the drug product for clinical use. In March 2022, the FDA cleared Sonnet’s Investigational New Drug (IND) application for
SON-1010. This allowed the Company to initiate a U.S. clinical trial (SB101) in oncology patients with solid tumors during the second
calendar quarter of 2022. In September 2021, the Company created a wholly-owned Australian subsidiary, SonnetBio Pty Ltd (“Subsidiary”),
for the purpose of conducting certain clinical trials. Sonnet received approval and initiated a clinical study (SB102) of SON-1010
in Australian healthy volunteers during the third calendar quarter of 2022. Interim safety and tolerability data from the SB101 and
SB102 studies were reported in April 2023. In January 2023, Sonnet announced a collaboration agreement with Roche for the clinical
evaluation of SON-1010 with atezolizumab (Tecentriq®). The companies have entered into a Master Clinical Trial and
Supply Agreement (MCSA), along with ancillary Quality and Safety Agreements, to study the safety and efficacy of the combination
of SON-1010 and atezolizumab in a platinum-resistant ovarian cancer (PROC) patient setting. Further, the companies will provide SON-1010
and atezolizumab, respectively, for use in the Phase 1b/Phase 2a combination safety, dose-escalation, and efficacy study (SB221).
That trial consists of a modified 3+3 dose-escalation design in Part 1 to establish the maximum tolerated dose (MTD) of SON-1010
with a fixed dose of atezolizumab. Clinical benefit in PROC will be confirmed in an expansion group to establish the recommended
Phase 2 dose (RP2D). Part 2 of the study will then investigate SON-1010 monotherapy or its use in combination with atezolizumab with
the standard of care (SOC) for PROC in a randomized comparison to show proof-of-concept (POC). As part of the Company’s ongoing
cost-cutting evaluations, all antiviral development with SON-1010 has been suspended. |
| |
● |
The
Company acquired the global development rights to a fully human version of Interleukin 6 (IL-6), in April 2020. We refer to this
candidate as SON-080, for its target indications of Chemotherapy-Induced Peripheral Neuropathy (CIPN) and Diabetic Peripheral Neuropathy
(DPN). Sonnet’s CIPN Phase 1b/2a clinical trial, SB221, is underway in Australia. Enrollment of the first portion
of SB211 study is nearing completion, which will position the DSMB to complete its review of the preliminary safety data during the
first calendar quarter of 2024. Pursuant to a license agreement that the Company entered into with New Life Therapeutics Pte., Ltd
(“New Life”) of Singapore in May 2021, Sonnet and New Life will be jointly responsible for developing SON-080 in DPN
with the objective of evaluating an ex-US pilot efficacy study after analyzing the CIPN safety data. |
| |
|
|
| |
● |
SON-1210
(IL12-FHAB-IL15), our lead bifunctional compound, combines the FHAB construct with single-chain IL-12 and fully
human Interleukin 15 (IL-15). This compound is being developed for solid tumor indications, including colorectal cancer. In February
2023, the Company announced the successful completion of two IND-enabling toxicology studies with SON-1210 in NHPs. Sonnet is prepared
to initiate the regulatory authorization process for SON-1210, pending the outcome of any partnering activity. |
In
our discovery pipeline, we are investigating:
| |
● |
SON-1410
(IL18-FHAB-IL12), a bifunctional combination of Interleukin 18 (IL-18) and IL-12, for melanoma and renal cancers. Cell
line development and process development are ongoing, with early experimental drug supply suitable for formulation and analytical
method development activities. After some development delays in 2023, activities will continue into 2024 with the potential to generate
a drug suitable for non-clinical studies and subsequent human studies. |
| |
● |
SON-3015
(anti-IL6- FHAB-anti-TGFβ), a bifunctional combination of anti-IL6 and anti-Tumor Growth Factor beta (TGFβ)
was being developed for tumor and bone metastases. The early-stage bifunctional drug has been generated and has been stored for future
use with in vivo mouse studies. Sonnet elected to place the SON-3015 development program on hold for expense reduction purposes. |
We
face numerous challenges and uncertainties with respect to the development and commercialization of our therapeutic compounds, including
our FHAB technology. Please see “Risk Factors” contained elsewhere in this prospectus, and the sections entitled
“Risk Factors” in the documents incorporated by reference into this prospectus.
Strategy
Our
goal is to rapidly advance our pipeline and leverage our therapeutic FHAB platform to become a leader in the discovery, development,
and commercialization of biologic drugs. Since its founding, Sonnet has remained focused on rapidly progressing pipeline candidates towards
the clinic, while also working to establish collaborations with suitable partners. As partnership conversations evolve, Sonnet intends
to prioritize its expense allocation on assets with the greatest strategic interest. To this end, Sonnet has reduced operating
expenses during fiscal year 2023 and intends to negotiate a licensing deal that will help fund future pipeline expansion. As one example
of a project in its early stages that was announced in October 2022, Janssen’s evaluation of three of our pipeline compounds, SON-1010,
SON-1210 and SON-1410, in combination with its cell therapy products remains ongoing.
FHAB
program advancement: SON-1010 has entered Phase 1b/2a clinical development to establish maximum tolerated dose (MTD) and to assess
clinical benefit in platinum-resistant ovarian cancer (PROC). Regarding our first bifunctional candidate, SON-1210, two IND-enabling
toxicology studies in NHPs have been successfully completed and the Company is prepared to initiate the regulatory authorization process,
pending the outcome of any partnering activity.
Progress
SON-080 into the next phase of clinical development: SON-080 is a fully human version of low dose IL-6 being studied for chemotherapy-induced
peripheral neuropathy (CIPN). IL-6 has successfully been studied in Phase 1 and Phase 2 clinical trials in cancer patients and we initiated
a pilot efficacy Phase 1b/2a study in CIPN patients during the second half of 2022.
Manufacturing
platform: Sonnet compounds are produced using an industry standard mammalian cell (Chinese Hamster Ovary (CHO)) host cell line that
allows for rapid scale-up and commercial manufacturing using state-of-the-art manufacturing processes and technologies. The mammalian
cell culture system enables glycosylation and a similar biological structure to the natural cytokines in vivo, which reduces the
chance of immunogenicity. The manufacture of cytokines for clinical applications, namely their production and purification, poses distinct
technical challenges. To this end, Sonnet has developed a proprietary continuous intensive perfusion manufacturing process, including
a proprietary ligand for efficient down-stream processing, as well as stable lyophilized formulations, for which we are seeking intellectual
property protection for certain of these manufacturing and downstream process development steps.
Regulatory
strategy: We believe that Sonnet’s drug candidates are significantly differentiated from existing therapies and represent potential
breakthroughs in biopharmaceutical drug development. We will endeavor to seek breakthrough therapy designations with regulatory agencies,
which could potentially lead to accelerated clinical development timelines.
Pipeline
licensing opportunities: We are pursuing partnering opportunities with leading biopharmaceutical companies for the development and
commercialization of our pipeline assets.
FHAB
technology expansion: Sonnet is exploring FHAB technology licenses with external partners interested in expanding its
therapeutic deployment, which we believe could lead to the platform’s application in other areas, such as vaccines, antibody drug
conjugates, and as a supplement to chimeric antigen receptor (CAR) T-cell technology in vivo. As soon as supportive data are available,
provisional patents will be filed to secure exclusivity with FHAB in these fields.
The
FHAB Technology
Our
proprietary FHAB technology was engineered to address several important shortcomings of existing approaches to biopharmaceutical
drug development. We designed the FHAB domain as a plug-and-play, modular construct for innovating new chemical entities that
is readily reconfigured for different therapeutic payloads. As is the case with all biologic drugs, dose level and frequency of administration
are critical variables that oftentimes present barriers to the development process. After injection, large molecule therapeutics, including
peptides, proteins, fusion proteins, antibodies, and the like, must remain intact and be capable of reaching their designated targets
inside the body, without exceeding specific toxicity thresholds. Finally, they must also be produced using commercially attractive means.
Sonnet’s
platform technology was designed to harness HSA as a therapeutic shuttling molecule. HSA is naturally present in the bloodstream and
the predominant protein in blood plasma. Albumin is a source of energy for inflamed, hypermetabolic tissues, including tumors. Due to
the active need for nutrients, cancer cells overexpress albumin-binding proteins such as the ‘Secreted Protein Acidic and Rich
in Cysteine’ (SPARC) and gp60 (albondin glycoprotein).
Pursuant
to a Discovery Collaboration Agreement, dated July 23, 2012, and to an Amendment of Discovery Collaboration Agreement, dated May 7, 2019
(together, the “Collaboration Agreement”), XOMA (US) LLC (“XOMA”) granted to Sonnet a non-exclusive, non-transferrable
license and/or right to use certain materials, technologies and information related to the discovery, optimization, and development of
antibodies and related proteins and to develop and commercialize products thereunder (each, a “Product”). The Collaboration
Agreement included a license to use a fully human bacteriophage library that was designed to generate fully human single-chain antibody
variable fragments (scFv) comprising a full repertoire of human heavy and light chains for use in panning biological sequences for specific
functions. Applying stringent criteria, Sonnet panned millions of scFv binders to HSA to generate Sonnet’s FHAB, which
binds to HSA, a globular protein having three major functional domains. It is known that albumin domains 1 and 3 are involved in the
binding to FcRn. This allowed Sonnet to select and characterize scFv binders that are specific to domain 2, a foundational aspect of
Sonnet’s FHAB platform.
Sonnet
is obligated to make contingent milestone payments to XOMA totaling $3.75 million on a Product-by-Product basis upon the achievement
of certain development and approval milestones related to a Product. To that point, the next projected clinical development milestone
of $750K is expected to be initiation of enrollment of a Product (i.e., SON-1010) in a Phase 2 Trial. Sonnet has also agreed to
pay XOMA low single-digit royalties on net sales of Products sold by Sonnet. Royalties on each Product are payable on a country-by-country
basis until the later of (i) twelve (12) years after the First Commercial Sale (as defined in the Collaboration Agreement), and (ii)
the date of expiration of the last valid claim in the last-to-expire of the issued patents covered by the Collaboration Agreement. In
addition, Sonnet has the right to reduce the rate of the royalty on a Product-by-Product basis by paying XOMA a specified amount. The
Collaboration Agreement may be terminated by either party for cause and contains customary indemnification provisions.
Sonnet’s
FHAB has demonstrated a high binding affinity to serum albumin across species (human, mouse and cynomolgus monkey), with little-to-no
immunogenicity, and retains the benefits of neonatal FcRn-mediated recycling of albumin for extending serum half-life. Unlike monoclonal
antibodies (mAbs), this binding occurs without invoking ADCC (antibody-dependent cellular cytotoxicity) or CDC (complement-dependent
cytotoxicity). The FHAB construct physically binds serum albumin (Figure 1) through an ionic, hydrophobic mechanism, which
we believe offers a distinct advantage over technologies that rely on chemical, covalent binding. Once broken, a covalent bond cannot
reform, whereas Sonnet’s FHAB is designed with the ability to bind, unbind and rebind to albumin in dynamic equilibrium.
As albumin also binds to the albumin receptors gp60 and SPARC, FHAB leverages innate biological mechanisms for targeted delivery
to and accumulation of the therapeutic payload in the tumor microenvironment.
Preclinical
radiolabeling studies have validated the tumor targeting attributes of the FHAB construct, where accumulation was demonstrated
in tumors compared to the same construct without FHAB, and was transient in liver, kidney, and other organs, as expected.
Importantly, radiolabeled FHAB also demonstrated measurable accumulation in the draining lymph nodes. These findings have
important implications for therapeutic applications of any mono- (ILx-FHAB) or bifunctional (ILx-FHAB-ILy) molecules
demonstrating enhanced tumor targeting and accumulation, as well as the potential for improved efficacy.
Another
unique advantage of Sonnet’s FHAB is its linker design (Figure 1) that is used for attaching one or two large molecule
therapeutic payloads for single or bifunctional activity. Our G4S (glycine, serine) peptide linkers are flexible, while being long enough
to prevent steric hindrance and can assume a rod-like configuration for enhanced penetration of tight tissue matrices. In addition to
maintaining distance between the therapeutic functional domains, Sonnet linkers are fully human and non-immunogenic across the linker
structure, including at the payload binding region. In bifunctional constructs, the orientation of the therapeutic payloads can be manipulated
to improve potential treatment effects.

As
a final key design component, FHAB is produced in mammalian cell culture, specifically Chinese Hamster Ovary (CHO) cells,
which enables glycosylation for reducing or potentially eliminating immunogenicity. Using CHO, we have created several different genetic
fusion constructs with various low molecular weight therapeutic proteins (e.g., recombinant cytokines such as IL-12, IL-15, IL-18, anti-IL-6,
and anti-TGFβ). Recombinant therapeutic proteins, including cytokines, have shown great therapeutic potential, but can lack tissue
specificity, which can lead to toxicity. Due to their small size (< 50 kDa), cytokines also suffer from a shorter circulation half-life
(minutes-to-hours versus 21 days for albumin) compared to monoclonal antibodies. In mouse and NHP models, FHAB-derived compounds
have demonstrated substantially greater serum half-lives, improved tissue accumulation, and have marked tumor reduction activity when
compared to their respective naked recombinant cytokines.
In
summary, our FHAB technology underpins a modular, versatile scaffold that can be customized to yield a broad array of multi-targeted
therapeutic candidates. Relative to existing albumin binding technologies, FHAB is differentiated by possessing a linear,
rod-like shape designed for better target tissue penetration, a fully human design to reduce immunogenicity, mammalian glycosylation,
and FcRn binding for longer serum half-life. Importantly, FHAB-derived therapeutics have the potential for targeted delivery
to tumor and lymphatic tissue, reduced toxicity, and wider therapeutic windows, with the added benefit of utilizing a tailored single-
or bifunctional mechanism of action.
Expanded
Applications of the FHAB Technology:
Immunotherapy:
We believe that our FHAB platform can innovate biologic drugs that target specific tissues while also increasing therapeutic
half-life. As the FHAB construct is designed to enable the simultaneous deployment of two synergistic immunotherapy compounds,
we envision a path to previously untapped immunotherapeutic advancements.
Drug
Conjugation: With the FHAB technology, various drug compounds can be linked to the FHAB scaffold in combinations
that extend beyond our first-wave pipeline of cytokines, which presents opportunities for development across myriad disease areas.
Vaccines:
Vaccine developers are seeking to improve vaccine efficiency by conjugating vaccines to natural carriers, such as albumin. We believe
the FHAB platform, with its modular scaffold structure, could be an efficient vehicle for delivering vaccines to lymph nodes,
improving penetration and presentation, and extending half-life.
CAR
T-cell Therapy: CAR T-cell therapy involves genetically modifying a patient’s own T cells to recognize cancer cells for more
effectively targeting and killing tumors. We believe targeted Sonnet constructs utilizing interleukins could be systemically co-administered
to enhance CAR T-cell efficacy.
Pipeline
Overview
The
following table summarizes information about pipeline programs where we have disclosed specific target indications:

SON-1010
IL-12
is a circulating cytokine that has been shown to exert multiple effects on innate and adaptive immunity. These immune functions are critical
in attacking cancer cells and pathogens. IL-12 is a heterodimeric cytokine produced by dendritic cells, monocytes, and macrophages, also
known as antigen presenting cells (APCs). IL-12 has been shown to induce interferon gamma (IFN-ɣ) secretion by T cells and natural
killer (NK) cells, promote the expansion and survival of activated T and NK cells, supplement the cytolytic activity of cytotoxic T cells,
support the differentiation of Th1 helper-effector cells and enhance antibody dependent cellular cytotoxicity (ADCC). IL-12 has also
been shown to stimulate in vitro antitumor activity of lymphocytes from patients with cancer and in vivo anti-tumor activity
in murine tumor models of melanoma, colon carcinoma, mammary carcinoma, and sarcoma.
Preclinical
Studies in Mice
Initially,
the murine version of SON-1010 (mIL12-FHAB) demonstrated a larger reduction of tumor growth preclinically compared to recombinant
mIL-12 without FHAB (naked/standalone IL-12) in a mouse model of melanoma. Figure 2, from this mouse melanoma study, illustrates
a 30-to-50-fold increase in tumor reduction with mIL12-FHAB compared to standalone mIL-12.
Furthermore,
in the same model, mIL12-FHAB accumulated in tumors in higher concentrations and remained in the serum, spleen, and tumor
significantly longer than mIL-12 without FHAB, potentially enabling less frequent administration and at lower doses.
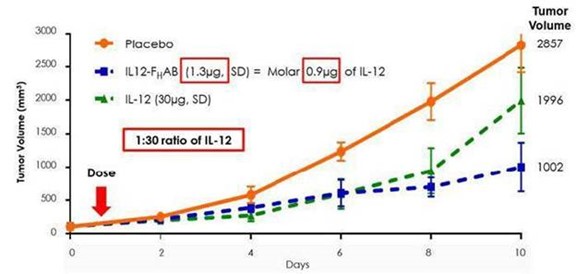
Figure
2: The molar equivalent for IL-12 (0.9µg) is IL12-FHAB (1.3 µg) and they have similar bioactivity in vitro; however,
in vivo, IL12- FHAB is approximately 30-fold more potent than IL-12 (at day 10, 1.3µg IL12-FHAB > IL-12
30µg).
In
another preclinical study using the B16F10 tumor model, mIL12-FHAB demonstrated an improved dose response versus recombinant
murine IL-12, along with increased survival duration (Figure 3 and Figure 4). Results from this study suggest that mIL12-FHAB
may have a greater effect on reducing tumor volume and extending survival versus standalone mIL-12.

Figure
3: Analysis of tumor volumes shows dose-dependent decreases in tumors in both mIL-12 and mIL12-FHAB-treated
mice, as compared to vehicle control. IL12-FHAB-treated mice showed statistically significant decreases in tumor volumes when
analyzed against equimolar-dosed, mIL-12-treated mice. Results suggest IL-12 anti-tumor activity is potentially enhanced with
the extension of serum half-life by FHAB linkage.
In
Figure 4, a Kaplan-Meier analysis was performed to compare survival between animals treated with either mIL12-FHAB or mIL-12.
These data illustrate a correlation between the decrease in tumor growth (Figure 3) and an increase in survival duration (Figure 4).
In this study, the slower growth of tumors in animals treated with mIL12-FHAB correlated with a longer survival time, as compared
to more rapid tumor growth observed with naked mIL-12 treatment. Survivability at the lowest doses of mIL12-FHAB (3µg)
was equivalent to the highest dose of mIL-12 (30µg). All doses of mIL12-FHAB showed a 50% survival increase over vehicle
at 14 and 17.5 days.

Figure
4: Kaplan-Meier evaluation of mouse B16F tumor survivability shows an increase in survival with IL12- FHAB treatment.
Doses of 10µg and 20µg of standalone mIL-12 exhibited 50% survival at 2 and 4 days over vehicle control (10 days). All doses
of IL12- FHAB showed 50% survival over vehicle at 14 and 17.5 days. Survivability at the lowest doses of IL12- FHAB
were equivalent to highest dose standalone IL-12
Nonhuman
Primate Studies of SON-1010
We
have completed in vitro pharmacology studies of affinity and binding kinetics that demonstrate species cross-reactivity of SON-1010
in serum albumin for hamster, rat, cynomolgus monkey and human. The results show that SON-1010 displays species specificity to cynomolgus
monkey and human subjects, which will guide species selection for further preclinical toxicology work. A humanized mouse model (SCID)
study designed to evaluate PK/PD and dose response is completed. This work informed our decision about dosing in a nonhuman primate (NHP)
study.
In
February 2021, we announced the successful completion of a NHP non-GLP repeat-dose toxicology study of SON-1010, the data from which
were used to inform the design of the cGLP toxicity study in preparation for IND submission. The objectives of the non-GLP study were
to evaluate the toxicity of SON-1010 in a repeat dose regimen at several dose levels and to gather critical data for the design of further
IND-enabling safety and toxicity studies. The study included both intravenous (IV) and SC routes of administration with a total of two
injections given 14 days apart. The highest dosage rate utilized in this study was greater than 50 times the anticipated clinical level
of exposure to patients. Study results included:
| | ● |
Repeat dosing by IV and
SC routes of administration was tolerated at both dose levels examined. As is typically observed with IL-12 administration, the white
blood cell count dropped, and liver enzymes (ALT and AST) were elevated. These were transient effects that returned to baseline within
7 days following the second dose. |
| | ● |
SON-1010-related changes
in the physiological observations, body weight, pathology, cytokines and immunophenotyping were seen, all of which were consistent
with those on-target effects previously observed in single dose studies. |
| | ● |
A significant increase
in IFN-γ levels, a key pleiotropic cytokine associated with anti-tumor activity, was observed following the initial dose of
SON-1010 with lower IFN-γ levels observed following the second dose. This trend follows the published data from other studies
of IL-12 in both humans and NHPs. Signs of cytokine imbalance, or uncontrolled increase of pro-inflammatory cytokines, including
TNF-α, IL-1β, and IL-6 were notably absent from all dose levels tested in the study. |
| | ● |
Pharmacokinetic analysis
indicated a mean serum half-life of approximately 40 hours in animals administered SON-1010 via SC injection. This is consistent
with data from the previously conducted dose escalation phase of the study, which demonstrates a substantial improvement in half-life
compared to the 13-19-hour half-life of naked, recombinant human IL-12. |
| | ● |
These results build on
those from the work with the B16F10 mouse model of melanoma, where the mouse version of SON-1010 showed a 20-fold reduction in the
dosage required to achieve a similar therapeutic effect compared to mouse IL-12. Taken together, we believe the observed extended
half-life, improved therapeutic window and reduced dosing requirement, made possible by Sonnet’s FHAB technology, represent
key advantages of SON-1010 as a potential immune oncology therapeutic. |
In
May 2021, we announced the successful completion of a cGLP repeat-dose study of SON-1010 in NHPs. The objectives of the study were to
evaluate the toxicity of SON-1010 in NHP using a subcutaneous (SC), repeat-dose regimen at three different dose levels versus untreated
controls and to evaluate the potential reversibility of any adverse findings. Study results included:
| |
● |
The
No Observed Adverse Event Level (NOAEL) following repeated administration in NHP was more than 50 times the anticipated equivalent
human clinical dose with no evidence of cytokine release syndrome. |
| |
● |
Pharmacokinetic
(PK) analysis of serum samples confirmed an enhanced profile of IL12-FHAB over recombinant human IL-12, with a half-life
around 40 hours in NHP. |
| |
● |
A
significant increase in IFN-γ, a key pleiotropic cytokine associated with anti-tumor mechanisms, was observed following dosing
with IL12-FHAB. |
| |
● |
SON-1010
related changes in clinical observations, body weight, clinical pathology, cytokines, and immunophenotyping were seen, all of which
were consistent with on-target effects previously observed in nonhuman primates. |
| |
● |
By
Day 38, all study subjects recovered to baseline (pre-study) laboratory values. |
| |
● |
Repeat
dosing administration was tolerated at all dose levels examined. |
Biodistribution
Studies
In
September 2023, we announced the completion of two independent in vivo proof-of-concept (POC) studies to show the biodistribution
of interleukin-FHAB molecules to the tumor microenvironment (TME), using labs with expertise in radiolabeling biologics and
in vivo biodistribution analysis. The labs employed different radiolabeling methodologies (99mTc or 89Zr)
for mIL-12 and mIL12-FHAB, either with or without a polyhistidine tag (His-Tag). The two studies were completed using the
B16F10 mouse melanoma model to measure the accumulation of radiolabeled product and tumor volume inhibition over various time points.
Both studies indicated that mIL12-FHAB had significantly higher tumor accumulation, 2.5-4.7 times higher on average at the
longer time points, and increased retention when compared to mIL-12. Accumulation was demonstrated in tumors compared to normal mice,
and was transient in liver, kidney, and other organs, as expected. Importantly, radiolabeled mIL12-FHAB also demonstrated
measurable accumulation in the draining lymph nodes. Overall, these findings have important implications for therapeutic applications
of any mono- (ILx-FHAB) or bi-functional (ILx-FHAB-ILy) molecules demonstrating enhanced tumor targeting and accumulation,
as well as the potential for improved efficacy that could lead to a variety of drug candidates.
Manufacturing
Development
Manufacturing
work on the master cell bank expressing SON-1010, formulation development, and process development activities have all been completed,
in addition to drug product presentation (liquid or lyophilized). Multiple cGMP drug product lots have been successfully manufactured
and provide inventory for ongoing clinical trials.
SON-1010
in the Clinic
We
initiated the first-in-human (FIH), Phase 1 trial (SB101) to assess the maximum tolerated dose (MTD) for adult patients with advanced
solid tumors and platinum-resistant ovarian cancer (PROC) in April 2022 and we presented initial data from the study at AACR in April
2023. More patients with PROC will be enrolled in the expansion portion of the study to confirm a recommended Phase 2 dose (RP2D). Of
the 15 patients from the first five cohorts of SB101 evaluable for follow-up at this latest cutoff, 9 had stable disease at the first
follow-up scan, 4 of which were already progressing at study entry. After four months follow-up, 5 of 14 patients remained stable at
the second scan, suggesting clinical benefit of SON-1010 in 36% of patients. The very first patient dosed, with an aggressive endometrial
sarcoma, had substantial tumor shrinkage with complete resolution of her ascites at one point, and has been clinically and radiographically
stable for over a year. Dosing in the first 3 cohorts was performed every 4 weeks but is now being done every 3 weeks in the new cohorts
to enhance safety at higher doses.
We
initiated a single-ascending dose (SAD) Phase 1 clinical study (SB102) in Australian healthy volunteers in July 2022 to carefully study
the PK and PD in preparation for potential combination studies. Data from the SB102 study were reported during the calendar first quarter
of 2023. Typical dose-related increases were seen with SON-1010 in the serum using a validated electrochemi-luminescence assay (Meso Scale
Diagnostics, LLC (MSD)) after SC administration. Drug levels peaked at about 11 hours with a geometric mean maximum concentration (Cmax)
of 29, 68, and 125 pg/mL for the 50, 100, and 150 ng/kg dose groups, respectively (Figure 5). The mean elimination half-life (T½)
after the 150 ng/kg dose of SON-1010 was 112 hours, compared to 12 hours for rhIL-12 given SC.

Figure
5: SON-1010 levels were assessed frequently after dosing, then followed on the days indicated for the rest of the SB102 study. The inset
shows the same data in more detail for the first week.
Among
the cytokine PD responses, the observed increases in IFN-ɣ were most pronounced and were dose-related, controlled, and prolonged.
SON-1010 induced IFN-ɣ in all active-drug subjects, which peaked at 24 to 48 hours then returned to baseline after 2 weeks (Figure
6). The IFN-ɣ geomean Cmax was 398, 384, and 666 pg/mL after 50, 100, or 150 ng/kg of SON-1010 respectively, while the
AUC0-48h was 6050, 10200, and 14600 h*pg/mL. Linear regression was used to predict the IFN-ɣ Cmax at higher
doses, which remains well within the range of safety. Low amounts of IL-10 were induced in a dose-dependent manner, which could also
have been in response to the increase in IFN-ɣ. There were small transient increases in IL-6, IL-8, and TNF-α after dosing
but no consistent pattern was seen with IL-1β, IL-2, or IL-4, and there was no evidence of cytokine release syndrome (CRS). Safety
was consistent with what has been reported previously; adverse events have generally been mild/moderate, transient in nature, and have
all been tolerable.

Figure
6: Cytokine levels were assessed frequently after dosing for PD, then followed on the days indicated for the rest of the SB102 study.
SON-1010
has been safe and tolerable at all doses tested to date. Adverse events have generally been mild/moderate and transient in nature, with
no study discontinuations for safety reasons. In addition, adverse effects have been less numerous and less intense with subsequent doses.
The geomean half-life (T½) of SON-1010 was 113 hours in SB101, compared to 12 hours for recombinant hIL-12 given SC
that has been observed in prior studies. Comparison of the PK curves between our two studies suggests that SON-1010 may be targeting
tumors, as the FHAB was designed to do. As with the healthy volunteers, cytokine analysis following each dose in the cancer
patients revealed a similar controlled and prolonged induction of IFN-γ that peaked at 24 to 48 hours and returned to baseline
after 2 to 4 weeks. A small increase in IL-10 was observed with each dose, as expected in response to IFN-γ. There was either minimal
or no signal for IL-1β, IL-6, IL-8, or TNF-α and no indication of any potential for cytokine release syndrome (CRS) at these
doses.
SON-080
for Chemotherapy Induced Peripheral Neuropathy
Through
our pipeline expansion efforts, we have identified IL-6 as a cytokine with important biological properties when delivered as a standalone
molecule. Our lead clinical stage asset, SON-080, is the native human version of IL-6 that is also manufactured in Chinese Hamster Ovary
(CHO) cells. A previous version of recombinant IL-6 has been studied in Phase 1 and Phase 2 clinical trials in cancer patients with thrombocytopenia
and in healthy volunteers. Sonnet’s comparable version will advance to the next stage of development in chemotherapy-induced peripheral
neuropathy (CIPN), a common side effect of treatment with antineoplastic agents in cancer. CIPN is a debilitating condition that manifests
itself as pain, numbness and tingling in the extremities. It has been reported in as many as 70% of patients undergoing specific cancer
regimens and is a leading cause of patients prematurely aborting chemotherapy. In animal experiments designed to replicate the clinical
symptoms of CIPN, recombinant IL-6 presented disease-modifying characteristics, including the potential to repair damaged nerves.
Based
on the preclinical work, we believe that SON-080 can potentially regenerate damaged nerves, thereby addressing not only the pain-related
symptoms, but also the profound discomfort and motor disability CIPN patients often experience. In the nervous system, IL-6 has exhibited
neurotrophic-like properties, inducing anti-apoptotic gene expression, protecting neurons from toxic injuries, and promoting nerve regeneration
and remyelination. IL-6 has demonstrated the potential to elicit nerve regrowth and to re-establish both normal nerve function (Figure
7) and sensations (Figure 6) in various preclinical models of CIPN induced by cisplatin, taxol, or vincristine. Activity from treatment
with SON-080 was also observed in preclinical models of type 2 diabetic neuropathy, outlining the potential for benefit in DPN, and other
diseases affecting the nervous system or other organs. This broad activity suggests that the SON-080 mechanism of action might not be
restricted to a given class of chemotherapeutic drugs and could elicit a universal neuroprotective-neurorestorative response. Additionally,
preclinical data point to the potential of SON-080 to elicit both preventive and curative activity in neuropathies (Figure 8). This introduces
the possibility of treating cancer survivors who still suffer from neuropathies, a population representing between 10% and 60% of the
14 million cancer survivors in the US.

Figure
7: Activity of IL-6 on neuropathy induced by taxol or cisplatin in rats measured at the histological (IENFD) or physiological (SNCV)
levels.

Figure
8: Data show preventive and curative activity potentiating restoration of normal sensitivity (here, using a behavioral response to hot
stimulus in cisplatin-induced peripheral neuropathy).
IL-6
has been studied in Phase 1 and Phase 2 studies in over 200 cancer patients with chemotherapy-induced thrombocytopenia. Trial enrollees
received SC doses ranging from 0.25 to 32 µg/kg, either daily or thrice weekly. In these trials, where solid tumor cancers were
present in more than 75% of the patients treated, the cumulative doses of IL-6 averaged in the 8000 μg range (122 - 54880 μg),
and the mean duration of treatment equaled 28 days. One of the trials covered six chemotherapy cycles, with an IL-6 treatment period
extending to 203 days. An exacerbation of either cancer or neuropathy was not observed in any of these trials.
The
MTD of SON-080 was determined in four studies by means of cohort dose escalations of sequential IL-6 dose groups utilizing established
common toxicity criteria. When administered daily, the MTD following daily SC injection was determined to be between 3 and 8 μg/kg;
when given 3 times per week, the MTD was estimated to be > 10 μg/kg. The most clinically relevant toxicities that defined the treatment-limiting
dose in these studies were flu-like symptoms and neurocortical toxicity, manifested by somnolence, restlessness, confusion, hallucination,
and disorientation. We anticipate using a dose of SON-080 that is 50-fold less than the prior IL-6 MTD and expect a more benign adverse
event profile going forward.
These
data form the basis for our clinical trials in CIPN, where dosing is expected to be significantly below MTD, as supported by the preclinical
studies. For comparison, our target dose will provide a cumulative dose that is 25 times below the mean cumulative dose reached for a
similar period of dosing. We also believe that SON-080 has significant potential for treating other neuropathies, including DPN, as well
as other diseases of the nervous system, and we are currently evaluating forward development paths for these opportunities. Sonnet initiated
an ex-US Phase 1b/2a pilot-scale efficacy study with SON-080 in CIPN in July 2022. The Data Safety Monitoring Board (DSMB) is scheduled
to review the initial safety findings after enrollment completes in Part 1, expected during the first calendar calendar quarter of 2024.
SON-080
for Diabetic Peripheral Neuropathy
In
addition to our CIPN program with SON-080, our DPN program may, subject to data collected from our planned CIPN studies with SON-080,
explore the clinical utility of IL-6 in diabetic peripheral neuropathy (DPN). DPN is currently diagnosed in 50%-80% of the diabetic patient
population. According to World Health Organization (WHO) projections, the prevalence of diabetes is estimated to exceed 350 million people
in 2030. Neuropathy is progressive and develops over the continuum of diabetes. The condition involves intractable pain with no obvious
origin, as well as non-pain-related symptoms such as loss of balance, lack of sensation, and autonomic dysfunctions, among others. These
deficits impair quality of life and lead to a reduction of life expectancy. Diabetic foot ulcers are a major cost associated with diabetic
medical care and are also directly linked to the development of DPN.
Notwithstanding
the seriousness of the condition, current treatments only address the pain component of DPN, leaving disease progression and non-pain-related
symptoms unaddressed. Furthermore, the few drugs currently used to reduce pain (i.e. Cymbalta, Lyrica, cannabinoids, opioids) are only
partially efficacious and are associated with major side effects, which typically delays their introduction into a patient’s care.
For these reasons, DPN remains a substantial unmet medical need with high commercial market potential.
Exercise
has long been recognized by WHO and caregivers as an effective means of treating and potentially preventing diabetes and several pilot
studies have provided evidence to support its role in improving DPN. However, a majority of diabetic patients are physically unable to
perform exercise. Regular exercise is known to improve diabetes-associated markers such as HbA1c and glucose homeostasis, to ameliorate
heart rate variability and to stimulate recovery of both nerve function and blood flow. Recent evidence demonstrates that IL-6 is released
during exercise and mediates some of the beneficial effects of physical activity. Sonnet has completed preclinical work in animal models
of DPN in which exogenous administration of IL-6 exhibited restorative activity in epidermal nerve density, nerve function, blood flow,
and reactions to painful or disturbing stimuli. In this context, SON-080 may become a future pivotal disease-modifying therapy for the
treatment of DPN.
In
vitro data on oligodendrocytes or organotypic cultures have shown that IL-6 potentially induces myelin gene expression by Schwann
cells or oligodendrocytes (Figure 9).

Figure
9: Illustration of survival (A) and differentiation of oligodendrocytes as assessed by myelin basic protein (MBP), proteolipid protein
(PLP) and its spliced variant expression (B).
Valerio
et al, Mol Cell Neurosci 21 (2002) 602-615.
Pizzi
et al, Mol Cell Neurosci 25 (2004) 301-311.
The
neuroprotective activity of IL-6 has been evaluated in various paradigms, including excitotoxicity. As well as protecting neurons, IL-6
potentially promotes axonal regeneration and restoration of functional synapses (Figure 10).

Figure
1011: Axonal regeneration activity in hemi-sectioned slices of the hippocampus (A), with increased expression of growth-associated protein
43 (GAP43) in injured slices but not in normal slices (NL) (B). Axonal regeneration activity across the lesion (C) and functional recovery
(D) of suppressed (A) excitatory postsynaptic potential (EPSP).Hakkoum et al, J Neurochem 100 (2007) 747-757.
The
activity of IL-6 in preclinical models of DPN has been evaluated by three independent laboratories. This work has shown that IL-6 exhibits
positive activity in neuropathy in a dose-dependent manner and may also help restore normal physiological parameters after neuropathy
is well established (i.e. four weeks after the induction of diabetes and consequential neuropathy). The beneficial activity is observed
on motor (Figure 11A) and sensory (Figure 11B) nerve function (conduction velocity), and behaviorally by measuring thermal (Figure 11C)
and tactile (Figure 11D) perceptions. In addition to the direct effects on myelin and axons previously observed in vitro, IL-6
has also been observed to have activity in restoring microvascular blood flow in the nerve in vivo (Figure 11E), which is a major
driver of diabetic neuropathies. Histological analyses of nerves in animals receiving preventive treatment with IL-6 during the development
of neuropathy suggest that IL-6 exhibits protective activity on myelin and may play a role in preserving nerve fiber integrity, as well
as nerve conduction velocity and the perception of sensations.

Figure
11: Curative treatment with IL-6 in rats with established diabetic neuropathy induced by streptozotocin.Cameron et al, Exp Neurol 207
(2007) 23-29.
Beyond
the oncology indication, 15 pilot studies totaling 167 subjects, including 27 patients with type 2 diabetes, were conducted by independent
academic groups not affiliated with Sonnet to evaluate the role of IL-6 in exercise and metabolism. The peer-reviewed results suggest
that low dose IL-6 mimics several beneficial aspects of exercise, including expression of anti-inflammatory molecules, increased lipid
metabolism, decreased insulin secretion, and activation of the STAT3 signaling pathway in muscle.
We
believe these data provide strong support for the clinical development of IL-6 in DPN. Through its mechanism of action and potential
disease modifying activity, low dose IL-6 may offer a therapeutic solution for neuropathic symptoms, as well as for cardiac autonomic
neuropathies (CAN), in diabetic patients. We intend to use data collected from our CIPN studies with SON-080 to inform our decision about
potential next development steps for SON-080 in DPN. Pursuant to the license agreement the Company entered with New Life of Singapore
in May 2021, we and New Life will be jointly responsible for developing SON-080 in DPN, with the objective of initiating an ex-US pilot
efficacy study in the second half of 2023.
SON-080:
New Life Therapeutics Agreement
In
May 2021, we announced the execution of a license agreement, described in detail below (the “New Life Agreement”) which resulted
in the out-license of our IL-6 (SON-080) asset for DPN to New Life of Singapore. The licensed territory includes the 10 ASEAN countries
of Singapore, Malaysia, Indonesia, Thailand, The Philippines, Cambodia, Brunei, Vietnam, Myanmar, and Lao PDR. In June and July of 2021,
we amended the New Life Agreement to make Sonnet BioTherapeutics, CH, SA (rather than Sonnet BioTherapeutics, Inc.) the party to the
New Life Agreement (First Amendment) and we also made Sonnet BioTherapeutics, Inc. the Guarantor of performance under the New Life Agreement
(Second Amendment), respectively. In addition to the initial $500,000 received by Sonnet upon signing of the LOI in August 2020, an additional
$500,000 non-refundable upfront payment was received by Sonnet upon execution of the New Life Agreement. According to the terms of the
New Life Agreement, Sonnet could receive a $1 million deferred license fee within 30 days of the achievement of an early commercial sales
milestone, a total of up to $19 million in milestone payments and a tiered royalty ranging from 12% to 30% on commercial sales. Sonnet
and New Life intend to evaluate the safety data from the CIPN study prior to making a determination about proceeding forward into the
clinical study of the DPN indication.
SON-1210
SON-1210,
our lead bifunctional construct, combines IL-12 and IL-15 conjugated to FHAB. These cytokines were selected based on synergistic
biologic activity. IL-15 acts through its specific receptor, IL15Rα, which is expressed on antigen-presenting dendritic cells (APC),
monocytes, and macrophages. In addition to the potential antitumor properties of IL-12 described above, we believe IL-15 can potentially
add the following complementary activity:
| |
● |
Induce
differentiation and proliferation of T, natural killer (NK) , and B cells |
| |
● |
Enhance
cytolytic activity of CD8+ T cells |
| |
● |
Induce
long-lasting CD8+ memory T cells enhancing immune surveillance against cancer for month/years |
| |
● |
Stimulate
differentiation and immunoglobulin synthesis by B cells |
| |
● |
Induce
maturation of dendritic cells |
| |
● |
Up
regulate IL-12β1 receptor expression |
We
have conducted a number of preclinical studies with the murine version of SON-1210 (mIL12-FHAB-hIL15). Mice injected once
with the doses indicated had suppressed tumor growth in the B16F10 melanoma model compared to controls (Figure 12). The combination of
IL-12 and IL-15 in cis with FHAB displayed synergistic activity beyond the tumor inhibition seen with mIL12-FHAB
(Figure 13). Overall, the reciprocal biologic activity of IL-12 and IL-15 suggests that:
| |
● |
IL-12:
Increases IL15Rα receptor, IFN-ɣ, NK/T cells, TH1 (tumor killing), and decreases Treg |
| |
● |
IL-15:
Increases IL12β 1 receptor, NK cells, CD8 memory and decreases apoptosis |

Figure
12: These data show an enhanced reduction in tumor growth with mIL12-FHAB-hIL15 compared to concomitantly administered,
naked mIL-12 and hIL-15 in a mouse model of melanoma.

Figure
13: The combination of IL-12 and IL-15 in cis with FHAB displayed synergistic activity, leading to improved tumor volume
reduction versus IL12-FHAB alone in a mouse model of melanoma.
In
February 2023, the Company announced the successful completion of two IND-enabling toxicology studies with SON-1210 in NHPs. A NHP non-GLP
dose escalation study of SON-1210 was completed in September 2022, and a GLP repeat dose NHP study was completed in the fourth calendar
quarter of 2022. cGMP manufacturing for bulk drug is complete, and a lyophilized formulation of drug product was manufactured in early
2023, to support the FIH clinical study. The initial tox material supported the non-GLP study, while the GLP study is being performed
on the same lot of GMP drug as intended for the Phase 1 clinical study. The regulatory authorization process for SON-1210 is scheduled
to commence pending the outcome of any partnering activity.
Discovery
Assets: SON-1410 (IL18-FHAB-IL12) and SON-3015 (Anti-IL6-FHAB-Anti-TGFβ)
In
August 2021, we announced the selection of a novel development candidate after completing comparative studies in a mouse melanoma model.
The candidate represents Sonnet’s second bifunctional compound integrating IL-12 and IL18 with the company’s FHAB
platform. The target indications for SON-1410 will be melanoma and renal cancers.
IL18-FHAB-IL12
showed statistically significant tumor size reduction in a mouse melanoma study compared with the placebo, as well as a dose response.
The data demonstrated:
| Compound | |
Day
0, Single Dose Tumor
@ 100 mm3 | |
Day
8 Tumor Volume (mm3 +/- SEM), N=8 | |
Day
8 Percentage Tumor Shrinkage | |
| Placebo | |
NA | |
1747 +/- 301 | |
| - | |
| IL18-FHAB-IL12 | |
1 µg | |
918 +/- 130 | |
| 47 | % |
| IL18-FHAB-IL12 | |
5 µg | |
619 +/- 141 | |
| 65 | % |
A
separate mouse study was also performed comparing the selected version of IL18-FHAB-IL12 with two other candidates, GMCSF-FHAB-IL18
and GMCSF-FHAB-IL12. The comparison data indicated significantly greater reduction in tumor volume, along with higher IFN-γ
levels and immune cell responses (NK, NKT, Th1, and cytotoxic CD8 T cells) using IL18-FHAB-IL12, compared with GMCSF-FHAB-IL12
or GMCSF-FHAB-IL18. Preclinical development continues for SON-1410 (IL18-FHAB-IL12), where cell line development
for GMP application is underway. After some delays in 2023, process development activities will continue into 2024, with the potential
to generate a drug suitable for GLP non-clinical studies in NHP’s, and subsequent human studies.
TGF-β/IL-6
biology is a strong predictor of overall survival in cancer, and combined targeting to suppress IL-6 and TGFβ signaling using SON-3015
may represent a promising strategy for treating tumor and bone metastases. TGFβ is released from degraded bone, and enhances IL-6
production, contributing to the vicious circle of bone metastasis. High FcRn expression in the bone environment would result in accumulation
in the bone of the dual construct anti-IL6-FHAB-anti-TGFβ, thereby potentially inhibiting or blocking bone metastases.
Sonnet has elected to place the SON-3015 development program on hold for expense reduction purposes.
We
face numerous challenges and uncertainties with respect to the development and commercialization of our therapeutic compounds, including
our FHAB technology. Please see “Risk Factors” contained elsewhere in this prospectus, and the sections entitled
“Risk Factors” in the documents incorporated by reference into this prospectus.
Competition
The
pharmaceutical and biotechnology industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis
on proprietary products. While we believe that our technology, development experience and scientific knowledge provide us with competitive
advantages, we face potential competition from many different sources, including large pharmaceutical and biotechnology companies, academic
institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and
establish collaborative arrangements for the research, development, manufacturing and commercialization of cancer immunotherapies. Any
product candidates that we successfully develop and commercialize will compete with new immunotherapies that may become available in
the future.
We
compete in the segments of the pharmaceutical, biotechnology and other related markets that develop immuno-oncology treatments. There
are many other companies that have commercialized and/or are developing immuno-oncology treatments for cancer including large pharmaceutical
and biotechnology companies, such as Amgen, AstraZeneca/MedImmune, Bristol-Myers Squibb, Merck, Novartis, Pfizer and Roche/Genentech.
We
face significant competition from pharmaceutical and biotechnology companies that target the use of specific cytokines or other large
molecules as immunomodulating therapies in the cancer setting. These generally include, single- or bi-specific antibodies, fusion proteins,
antibody drug conjugates and targeted vaccines.
With
respect to our lead product candidate, SON-080, we are aware of other companies developing products to treat CIPN, including but not
limited to Aphios Corporation, Asahi Kasei Corporation, MundiPharma EDO and Regenacy Pharmaceuticals, Inc; however, we believe we are
the only company studying the use of a disease-modifying cytokine for the indication.
With
respect to our first FHAB-derived candidate, SON-1010, we are aware of other competing IL-12 programs, which include, but
are not limited to those being developed by Celsion Corporation, Eli Lilly, Inovio Pharmaceuticals, Inc., Intrexon Corporation, Codiak
Biosciences, Xiolio Therapeutics, Werewolf Therapeutics, Dragonfly Therapeutics and OncoSec Medical. We believe that our FHAB
integrated IL-12 is tumor-targeted with an enhanced PK profile that differentiates it from the competition.
With
respect to our earlier stage pipeline FHAB product candidates SON-1210, SON-2014 and SON-3105, we are not aware of any other
competing companies working on these specific bifunctional programs.
Many
of the companies against which we are competing or against which we may compete in the future have significantly greater financial resources
and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals
and marketing approved drugs than we do. Mergers and acquisitions in the pharmaceutical, biotechnology and diagnostic industries may
result in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also
prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors
also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites
and enrolling subjects for our clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
We
could see a reduction or elimination of our commercial opportunity if our competitors develop and commercialize products that are safer,
more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we or our collaborators
may develop. Our competitors also may obtain FDA or foreign regulatory approval for their products more rapidly than we may obtain approval
for ours, which could result in our competitors establishing a strong market position before we or our collaborators are able to enter
the market. The key competitive factors affecting the success of all our product candidates, if approved, are likely to be their efficacy,
safety, convenience, price, the effectiveness of companion diagnostics, if required, the level of biosimilar or generic competition and
the availability of reimbursement from government and other third-party payors.
Manufacturing
We
rely on contract development and manufacturing organizations, or CDMOs, to produce our drug candidates in accordance with the FDA’s
current Good Manufacturing Practices, or cGMP, regulations for use in our clinical trials. The manufacture of biopharmaceuticals is subject
to extensive cGMP regulations, which impose various procedural and documentation requirements and govern all areas of record keeping,
production processes and controls, personnel and quality control. Our pipeline molecules are manufactured using the standard industrial
Chinese Hamster Ovary (CHO) platform with common bio-chemical engineering from readily available raw materials.
To
meet our projected needs for clinical supplies to support our activities through regulatory approval and commercial manufacturing, the
one of the CDMOs with whom we currently work has increased their scale of production, and is building a cGMP manufacturing site in the
United States, available by Q3, 2024. The landscape for CDMOs is strong and there are multiple potential sources for contract manufacturing.
We have not yet engaged alternate suppliers since our current CDMO is able to scale production and continues to successfully manufacture
Sonnet’s pipeline. Our relationships with CDMOs are managed by internal personnel with extensive experience in pharmaceutical development
and manufacturing.
License
and Other Commercial Arrangements
Janssen
Pharmaceuticals (Johnson & Johnson)
In
October 2022, Sonnet announced a collaboration agreement with Janssen Biotech, Inc. (Janssen), one of the Janssen Pharmaceutical Companies
of Johnson & Johnson, where in vitro and in vivo efficacy of SON-1010 (IL12-FHAB), SON-1210 (IL12-FHAB-IL15)
and SON-1410 (IL18-FHAB-IL12) will be evaluated in combination with certain Janssen proprietary cell therapy assets. The agreement
was facilitated by Johnson & Johnson Innovation. Under the terms of the agreement, Sonnet shall supply the three referenced compounds
for use in head-to-head in vitro and in vivo efficacy studies. If successful and subject to provisions of the agreement,
Janssen could exercise its option and Sonnet could then seek a license and/or an expanded collaboration.
Roche
In
January 2023, Sonnet announced a collaboration agreement with Roche for the clinical evaluation of SON-1010 with atezolizumab (Tecentriq®).
The companies have entered into a Master Clinical Trial and Supply Agreement (“MCSA”), along with ancillary Quality and Safety
Agreements, to study the safety and efficacy of the combination of SON-1010 and atezolizumab in a platinum-resistant ovarian cancer (“PROC”)
patient setting. Further, the companies will provide SON-1010 and atezolizumab, respectively, for use in the Phase 1b/Phase 2a combination
safety, dose-escalation, and efficacy study (SB221).
New
Life
In
May 2021, the Company entered into a License Agreement (the “New Life Agreement”) with New Life Therapeutics PTE, LTD (“New
Life”). Under the New Life Agreement, the Company granted New Life an exclusive license (with the right to sublicense) to develop
and commercialize pharmaceutical preparations containing a specific recombinant human IL-6, SON-080 (the “Compound”) (such
preparations, the “Products”) for the prevention, treatment or palliation of diabetic peripheral neuropathy in humans (the
“DPN Field”) in Malaysia, Singapore, Indonesia, Thailand, Philippines, Vietnam, Brunei, Myanmar, Lao PDR and Cambodia (the
“Exclusive Territory”). New Life may exercise an option to expand (1) the field of the exclusive license to include the prevention,
treatment or palliation of chemotherapy-induced peripheral neuropathy in humans (the “CIPN Field”), which option is non-exclusive
and will expire on December 31, 2021; and/or (2) the territorial scope of the license to include the People’s Republic of China,
Hong Kong and/or India, which option is exclusive and will also expire on December 31, 2021. If these options are exercised, the terms
of the CIPN Field and the territory expansion will be negotiated by the parties. In June and July of 2021, we amended the New Life Agreement
to make Sonnet BioTherapeutics CH SA (rather than Sonnet BioTherapeutics, Inc.) the party to the New Life Agreement (First Amendment)
and we also made Sonnet BioTherapeutics, Inc. the Guarantor of performance under the New Life Agreement (Second Amendment), respectively.
The
Company will retain all rights to manufacture Compounds and Products anywhere in the world. The Company and New Life shall enter into
a follow-on supply agreement pursuant to which the Company shall supply to New Life Products for development and commercialization thereof
in the DPN Field (and the CIPN Field, if applicable) in the Exclusive Territory on terms to be negotiated by the parties. The Company
will also assist in transferring certain preclinical and clinical development know-how that is instrumental in New Life’s ability
to benefit from the license.
New
Life will bear the cost of, and be responsible for, among other things, conducting clinical studies and additional non-clinical studies
and other developmental and regulatory activities for and commercializing Products in the DPN Field (and the CIPN Field, if applicable)
in the Exclusive Territory.
New
Life paid the Company a $0.5 million non-refundable upfront cash payment in August 2020 upon executing a letter of intent to negotiate
a license agreement and a $0.5 million non-refundable upfront cash payment in June 2021 in connection with the execution of the New Life
Agreement. New Life is also obligated to pay a non-refundable deferred license fee of an additional $1.0 million at the time of the satisfaction
of certain milestones, as well as potential additional milestone payments to the Company of up to $19.0 million subject to the achievement
of certain development and commercialization milestones. In addition, during the Royalty Term (as defined below), New Life is obligated
to pay the Company tiered double digit royalties ranging from 12% to 30% based on annual net sales of Products in the Exclusive Territory.
The “Royalty Term” means, on a Product-by-Product and a country-by-country basis in the Exclusive Territory, the period commencing
on the date of the first commercial sale (subject to certain conditions) of such Product in such country in the Exclusive Territory and
continuing until New Life ceases commercialization of such Product in the DPN Field (or CIPN Field, if applicable).
The
New Life Agreement will remain in effect on a Product-by-Product, country-by-country basis and will expire upon the expiration of the
Royalty Term for the last-to-expire Product in the last-to-expire country, subject to (i) each party’s early termination rights
including for material breach or insolvency or bankruptcy of the other party and (ii) the Company’s Buy Back Right and New Life’s
Give Back Right (as defined below).
In
addition, New Life granted to the Company an exclusive option to buy back the rights granted by the Company to New Life and the Company
granted New Life the right to give back the rights with respect to Products in the DPN Field and/or the CIPN Field (if applicable) in
one or more countries in the Exclusive Territory on terms to be agreed upon, which options will expire upon the initiation of a Phase
III Trial for the applicable Product.
XOMA
Sonnet
(as successor-in-interest to Oncobiologics, Inc. (“Oncobiologics”), after Oncobiologics spun-off certain assets into Sonnet
and concurrently distributed all of its shares in Sonnet on a pro rata basis to Oncobiologics’s stockholders on April 6, 2015)
and XOMA (US) LLC (“XOMA”) are party to a Discovery Collaboration Agreement, dated July 23, 2012 and an Amendment of Discovery
Collaboration Agreement, dated May 7, 2019 (together, the “Collaboration Agreement”) pursuant to which XOMA granted to Sonnet
a non-exclusive, non-transferrable license and/or right to use certain materials, technologies and related information related to discovery,
optimization and development of antibodies and related proteins and to develop and commercialize products thereunder (each, a “Product”).
Sonnet is obligated to make contingent milestone payments to XOMA totaling $3.75 million on a Product-by-Product basis upon the achievement
of certain development and approval milestones related to a Product. To that point, the next projected clinical development milestone
of $750K is expected to be initiation of enrollment of a Product (i.e., SON-1010) in a Phase 2 Trial. Sonnet has also agreed to
pay XOMA low single-digit royalties on net sales of Products sold by Sonnet. Royalties on each Product are payable on a country-by-country
basis until the later of (i) a specified period of time after the First Commercial Sale (as defined in the Collaboration Agreement),
and (ii) the date of expiration of the last valid claim in the last-to-expire of the issued patents covered by the Collaboration Agreement.
In addition, Sonnet has the right to reduce the rate of the royalty on a Product-by-Product basis by paying XOMA a specified amount.
The Collaboration Agreement may be terminated by either party for cause and contains customary indemnification provisions.
ARES
On
August 28, 2015, Relief, now a wholly owned subsidiary of Sonnet, signed a License Agreement (the “ARES License Agreement”)
with Ares Trading, a wholly owned subsidiary of Merck KGaA (“ARES”). Under the terms of the ARES License Agreement, ARES
has granted the Company a sublicensable, exclusive, worldwide, royalty-bearing license on proprietary patents to research, develop, use
and commercialize products (each, a “Product”) using atexakin alfa (“Atexakin”), a low dose formulation of human
IL-6 in peripheral neuropathies and vascular complications. Three patents are included in the ARES License Agreement that protect the
use of Atexakin to treat i) diabetic neuropathy, ii) chemotherapy-induced peripheral neuropathy and iii) vascular complications.
Pursuant
to the ARES License Agreement, we will pay ARES high single-digit royalties on net sales of Products sold by the Company. Royalties are
payable on a Product-by-Product and country-by-country basis until the later of (i) a specified period of time after the First Commercial
Sale (as defined in the ARES License Agreement) in such country, and (ii) the last date on which such product is covered by a valid claim
in such country. If a Product is not covered by a valid claim in a country or such valid claim has expired or been invalidated before
the twelfth (12th) anniversary of the date of the First Commercial Sale of such Product in such country, then the royalty rate will be
reduced by fifty percent (50%). We will also pay ARES a sublicensing fee that is a percentage of the proceeds received from a sublicensing
event (“Sublicensing Receipts”) using a sliding scale (which percentage decreases at later stages of clinical development
at which the sublicensing event occurs) that starts in the low double digits and decreases to the high single digits. The ARES License
Agreement may be terminated by the Company for convenience at any time or by either party upon a breach by the other party. The License
agreement contains customary indemnification provisions.
The
Ares License Agreement was amended effective November 1, 2021, in order to clarify the application of some of the terms and conditions
contained therein related to sublicensing. In particular:
| |
● |
Sonnet
is now authorized to grant sublicenses to third parties without the prior written consent of ARES, providing that the financial condition
of any such sublicenses reflects fair market value as determined by Sonnet in good faith. |
| |
● |
Because
the initial conditions by which Sonnet would remunerate ARES out of Sublicensing Receipts were unclear, the ARES License Agreement
was clarified such that Sonnet will now have to pay ARES a percentage of all Sublicensing Receipts in case the relevant sublicense
agreement is signed before or after completion of the first Phase 1 clinical trial (as opposed to payment only in case the relevant
sublicense agreement is signed after completion of the first Phase 1 clinical trial, as was set in the original ARES License Agreement). |
| |
● |
It
was agreed that the foregoing clarification would only apply to future sublicensing agreements, and with respect to the royalties
(but not the milestone payments) that may be generated from the New Life Agreement. |
Intellectual
Property
With
respect to our patent portfolio, we have four issued patents (U.S., Japan, New Zealand and Russia), and we have filed patent applications
directed to numerous fusion proteins that include the Fully Human Albumin Binding domain (FHAB). If granted, the resulting
patents would expire on dates ranging from 2038 to 2041, subject to patent term extensions under certain circumstances. The patent application
filings include:
●
National filings corresponding to WO/2018/151868 - This application is directed to fully human “Albumin Binding Domain (FHAB)
Fusion Proteins,” including fusion proteins with scFv’s (e.g., anti-TGFβ, PD-L1, TNF, IL-1, IL-6, IL-8, etc.),
fusion proteins with cytokines (e.g., IL-2-FHAB, IL-12-FHAB, IL-15-FHAB, IL-7-FHAB,
etc.) and combinations of two cytokines, such as IL-12-FHAB-IL-15, GM-CSF-FHAB-IL-18, and IL-18-FHAB-IL-12;
and methods of treatments using such FHAB fusion proteins. A patent was issued in the United States on June 8, 2021, as U.S.
Patent No. 11,028,166. A patent was issued in Japan on December 23, 2022, as Japanese Patent No. 7200138. A patent was issued in Russia
on December 21, 2022, as Russian Patent No. 2786444. A patent was issued in New Zealand on October 3, 2023, as New Zealand Patent No.
756674. U.S. Patent No. 11,028,166 is currently estimated to expire on March 26, 2039, while Japanese Patent No. 7200138, Russian Patent
No. 2786444, and New Zealand Patent No. 756674 are estimated to expire on February 20, 2038. Applications are also pending in Australia,
Brazil, Canada, China, Europe, Hong Kong, and India. Continuation and divisional applications are pending in the United States and Japan,
respectively.
●
U.S. Patent No. 11,028,166 and the PCT patent application (PCT/US2018/00085), titled “Albumin Binding Domain Fusion Proteins”
originally received an application filing date of February 20, 2018, which is four days after the one-year anniversary of the filing
date of U.S. provisional patent applications U.S. 62/459,975 and U.S. 62/459,981 to which both the U.S. patent and the PCT patent application
claim a priority benefit. A request to restore the priority benefit to the filing date of U.S. provisional patent applications U.S. 62/459,975
and U.S. 62/459,981 was granted for the U.S. patent and PCT patent application. Subsequently, national phase patent applications were
filed from the PCT patent application in Australia, Brazil, Canada, China, Europe, India, Japan, New Zealand and Russia. However, due
to differences in the patent laws in these jurisdictions, the priority claims to U.S. 62/459,975 and U.S. 62/459,981 have thus far only
been accepted in Australia, Europe, India, Japan, New Zealand, and Russia.
●
US provisional application directed to anti-IL6-FHAB fusion proteins, including anti-IL6-FHAB, anti-IL6-FHAB-anti-TGFβ,
and anti-IL6-FHAB-anti-IL8 fusion proteins; and methods of treatments using such fusion proteins was re-filed as US 63/245,702
on September 22, 2021. However, due in large part to scientific challenges, the supportive data was not obtained within the one-year
period after filing the provisional patent, and therefore, the patent was abandoned.
●
US provisional application directed to Antigen/Albumin Binding Domain Conjugates, and methods of treatments using such conjugates was
re-filed as US 63/187,278 on May 11, 2021. Data in support of the provisional patent claims was not generated, and therefore, this patent
was abandoned.
●
US provisional application directed to Method of Treating Age-Related Frailty with Interleukin-6 filed June 4, 2021, as Application no.
63/197,097 and converted to a PCT patent on June 3, 2022.
●
US provisional application directed to Antibody-Based Drug Conjugates filed December 7, 2021, as Application no. 63/286,996. This provisional
patent was abandoned due to insufficient supportive data within the one-year timeframe.
●
US provisional application directed to IL-12-Albumin-Binding Domain Fusion Protein Formulations and Methods of Use Thereof filed on May
27, 2022, as Application no. 63/346,368. This provisional patent was converted to a PCT application (PCT/US2023/067566) on May 26, 2023.
●
US provisional application directed to Low Dose IL-6 Formulations and Methods of Use Thereof filed on March 14, 2023, as Application
no. 63/490,202.
●
US provisional application directed to Low Dose IL-6 Formulations and Methods of Use Thereof filed on September 30, 2022, as Application
no. 63/377,971. This provisional patent was converted to a PCT application (PCT/US2023/075593) on September 29, 2023.
●
US provisional application directed to Methods for the Treatment of Cancer with Recombinant IL-12 Albumin Binding Domain Fusion Proteins
filed on November 2, 2022, as Application no. 63/421,846. This provisional patent was converted to a PCT application (PCT/US2023/078366)
on November 1, 2023.
With
respect to our trademark portfolio, we received international registrational approval with the World Intellectual Property Office (WIPO)
for the Sonnet BioTherapeutics and FHAB marks, each having an Effective Date of Sept. 17, 2020. Further, both marks were published
by the European Union Intellectual Property Office (EUIPO), having Effective Dates of Nov. 30, 2020 and Dec. 6, 2020, respectively. In
2021, the USPTO issued Notices of Allowance for both marks, indicating that both applications have successfully completed the opposition
period and have matured to registration with the submission acceptable Statements of Use. To that end, the USPTO issued a Notice of Allowance
of the Statement of Use for each of the Sonnet BioTherapeutics and FHAB applications and the Sonnet BioTherapeutics mark already
received a Certificate of Registration under Registration no. 6,790,475.
●
The Switzerland Trademark Office granted protection to the Sonnet BioTherapeutics and FHAB marks on Sept. 14, 2021, and Oct. 26, 2021,
respectively, and are protected under International Trademark Registration nos. 1558330 and 1558471.
●
The Canadian Intellectual Property Office granted protection to the Sonnet BioTherapeutics mark on June 8, 2022 and is protected under
International Trademark Registration no. 1558330 while the FHAB mark is protected under International Trademark Registration no. 15584471,
for which the 18-month opposition period began on Nov. 16, 2022.
●
In addition to Switzerland and Canada, the Sonnet BioTherapeutics mark was also granted protection in Australia, European Union, Japan,
Mexico, South Korea and the United Kingdom, in each case having a Registration no. of 1558330, an Effective Registration date of Sept.
17, 2020 and a renewal date of Sept. 17, 2030. Likewise, the FHAB mark was granted protection in Australia, China, European Union, Japan,
Mexico, South Korea and the United Kingdom, in each case having a Registration no. of 1558471, a Granted Protection Date of Sept. 17,
2020 and renewal date of Sept. 17, 2030.
●
Although the Sonnet BioTherapeutics mark was initially rejected in China due to potential non-use claims directed to certain competing
companies, our law firm is quite confident that since the initial Class 42 rejection was successfully cancelled, two new trademark applications
for this same mark were also registered and/or published in 2021 could also be overcome; however, we won’t be able to initiate
non-use cancellation filings against these marks until 2025, which is the anticipated timeframe by which these pending class 42 applications
are likely to become registered in China.
Employees
As
of September 30, 2023, we had 12 full-time employees. None of our employees are represented by a labor union or covered by a collective
bargaining agreement, and we believe our relationship with our employees is good. Additionally, we utilize independent contractors and
other third parties to assist with various aspects of its business.
Government
Regulation
The
research, development, testing, manufacture, quality control, approval, packaging, storage, recordkeeping, labeling, advertising, promotion,
distribution, marketing, post-approval monitoring and reporting, and import and export of pharmaceutical products, including biological
products, are extensively regulated by government authorities in the United States, at the federal, state and local level, and other
countries and jurisdictions. Some jurisdictions also regulate the pricing of pharmaceutical products. The processes for obtaining marketing
approvals in the United States and in foreign countries and jurisdictions, along with subsequent compliance with applicable statutes
and regulations and other regulatory authorities, require the expenditure of substantial time and financial resources.
Licensure
and Regulation of Biologics in the United States
In
the United States, biological products, or biologics, are regulated under the Public Health Service Act, or PHSA, and the Federal Food,
Drug, and Cosmetic Act, or FDCA, and their implementing regulations. The failure to comply with the applicable requirements at any time
during the product development process may subject an applicant to delays in the conduct of a study, regulatory review and approval,
and/or administrative or judicial sanctions. These sanctions may include, without limitation, the FDA’s refusal to allow an applicant
to proceed with clinical testing, refusal to approve pending applications, license suspension or revocation, withdrawal of an approval,
product recalls, product seizures, suspension of production or distribution, injunctions, fines, investigations and civil and criminal
penalties. Biological product candidates must be granted a biological license by the FDA before they may be legally marketed in the United
States.
The
process required by the FDA to obtain a biological license in the United States generally involves the following:
●
Completion of extensive nonclinical, or preclinical, laboratory tests and preclinical animal trials and applicable requirements for the
humane use of laboratory animals and formulation studies in accordance with applicable regulations, including good laboratory practices,
or GLPs;
●
Submission to the FDA of an investigational new drug, or IND, application prior to initiation of any human clinical trials. Permission
to proceed must be received before the beginning of such trials;
●
Performance of adequate and well-controlled human clinical trials to establish the safety, potency and purity of the product candidate
for each proposed indication, in accordance with the FDA’s regulation generally referred to as the good clinical practices, or
GCP and any additional requirements for the protection of human research subjects and their health information, to establish the safety
and efficacy of the proposed biological product for its intended use. The FDA may also impose clinical holds on biological product candidate
at any time before or during our clinical trials due to safety concerns or non-compliance. If the FDA imposes a clinical hold, trials
may not recommence without FDA authorization and then only under terms authorized by the FDA;
●
Preparation and submission to the FDA of a Biologic License Application, or BLA, for a biologic product requesting marketing for one
or more proposed indications, including submission of detailed information on the manufacture and composition of the product in clinical
development and proposed labeling;
●
Review of the product by an FDA advisory committee, as determined by the FDA review division;
●
Satisfactory completion of one or more FDA inspections of the manufacturing facility or facilities, including those of third parties,
at which the product, or components thereof, are produced to assess compliance with current Good Manufacturing Practices, or cGMP, requirements
and to assure that the facilities, methods and controls are adequate to preserve the product’s identity, strength, quality and
purity;
●
Satisfactory completion of one or more FDA audits of the clinical study sites to assure compliance with GCPs, and the integrity of clinical
data in support of the BLA;
●
Payment of user fees and securing FDA approval of the BLA and licensure of the new biologic product;
●
Compliance with any post-approval requirements, including the potential requirement to implement a Risk Evaluation and Mitigation Strategy,
or REMS, and any post-approval studies required by the FDA.
Nonclinical
Studies and Investigational New Drug Application
Each
product candidate must undergo nonclinical testing before testing in humans. These tests include laboratory evaluations of product chemistry,
formulation and stability, as well as animal studies to evaluate the potential for activity and toxicity and must be conducted in compliance
with applicable regulations. The results of the nonclinical tests, together with manufacturing information and analytical data, are submitted
to the FDA as part of an IND application. The IND automatically becomes effective 30 days after receipt by the FDA, unless before that
time the FDA raises concerns or questions about the product or conduct of the proposed clinical trial, including concerns that human
research subjects will be exposed to unreasonable health risks. In that case, the IND sponsor and the FDA must resolve any outstanding
FDA concerns before the clinical trial can begin.
Submission
of the IND may result in the FDA not allowing the trial to commence or on the terms originally specified by the sponsor in the IND. If
the FDA raises concerns or questions, it may choose to impose clinical holds on biological product candidates at any time before or during
clinical trials due to safety concerns or non-compliance. If the FDA imposes a clinical hold, trials may not recommence without FDA authorization
and only under terms authorized by the FDA.
Human
Clinical Trials in Support of a BLA
Clinical
trials involve the administration of the investigational product candidate to healthy volunteers or patients with the disease to be treated
under the supervision of a qualified principal investigator in accordance with GCP requirements. A protocol for each clinical trial and
any subsequent protocol amendments must be submitted to the FDA as part of the IND. A sponsor who wishes to conduct a clinical trial
outside the United States may, but need not, obtain FDA authorization to conduct the clinical trial under an IND. If a foreign clinical
trial is not conducted under an IND, the sponsor may submit data from the clinical trial to the FDA in support of the BLA so long as
the clinical trial is well-designed and well-conducted in accordance with GCP, including review and approval by an independent ethics
committee, and the FDA is able to validate the study data through an onsite inspection, if necessary.
Further,
each clinical trial must be reviewed and approved by an institutional review board, or IRB, either centrally or individually at each
institution at which the clinical trial will be conducted or, for trials conducted outside of the United States, by an independent ethics
committee referred to above. The IRB will consider, among other things, clinical trial design, patient informed consent, ethical factors
and the safety of human subjects. An IRB must operate in compliance with FDA regulations. The FDA, IRB, or the clinical trial sponsor
may suspend or discontinue a clinical trial at any time for various reasons, including a finding that the clinical trial is not being
conducted in accordance with FDA requirements or the subjects or patients are being exposed to an unacceptable health risk. Clinical
testing also must satisfy extensive GCP rules and the requirements for informed consent. Additionally, some clinical trials are overseen
by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee.
This group may recommend continuation of the study as planned, changes in study conduct, or cessation of the study at designated check
points based on access to certain data from the study.
Clinical
trials typically are conducted in three sequential phases that may overlap or be combined. Additional studies may be required after approval.
●
Phase 1: the biological product candidate is initially introduced into healthy human volunteers and tested for safety. In the
case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer
to healthy volunteers, the initial human testing is often conducted in patients, such as cancer patients.
●
Phase 2: the biological product candidate is evaluated in a limited patient population to identify possible adverse effects and
safety risks, preliminary evaluate the efficacy of the product for specific targeted diseases and determine dosage tolerance, optimal
dosage and dosing schedule.
●
Phase 3: Clinical trials are undertaken to further evaluate dosage, clinical efficacy, potency and safety in an expanded patient
population and geographically dispersed clinical study sites. These trials are intended to establish the overall risk/benefit ratio of
the product and provide adequate basis for product labelling.
●
Phase 4: post-approval clinical trials, or Phase 4 clinical trials, may be conducted after initial marketing approval. They provide
additional experience for the treatment of patients in the intended therapeutic indication, particularly for long-term safety follow-up.
If the FDA approves a product while a company has ongoing clinical trials that were not necessary for approval, a company may be able
to use the data from these clinical trials to meet all or part of any Phase 4 clinical trial requirement or to request a change in the
product labeling. Failure to exhibit due diligence with regard to conducting required Phase 4 clinical trials could result in withdrawal
of approval for products.
Compliance
with cGMP Requirements
Before
approving a BLA, the FDA will typically inspect the facility(ies) where the product is manufactured to ensure full compliance of the
manufacturing processes and facilities with cGMP requirements and consistent production with required specifications. Manufacturers and
others involved in the manufacture and distribution of products must also register their establishments with the FDA and certain state
agencies. Both domestic and foreign manufacturing establishments must register and provide additional information to the FDA upon their
initial participation in the manufacturing process. Any product manufactured by or imported from a facility that has not registered is
deemed misbranded under the FDCA. Establishments may be subject to periodic unannounced inspections by government authorities. Manufacturers
may have to provide records regarding their establishments.
Review
and Approval of a BLA
Results
of product candidate development, nonclinical testing and clinical trials are submitted to the FDA as part of a BLA requesting a license
to market the product. The BLA must contain extensive and detailed information on the manufacturing and composition of the product and
proposed labeling as well as payment of a user fee. The FDA has 60 days after submission of the application to conduct an initial review
to determine whether the BLA is sufficient to accept for filing. Once the submission has been accepted for filing, the FDA begins its
in-depth review. The FDA has twelve months in which to complete its initial review of a standard application (or six months for a priority
review) and respond to the applicant. The FDA does not always meet its goal dates and the review process may be significantly extended
by FDA requests for additional information or clarification. The review process and the goal date may be extended by three months if
the FDA requests or if the applicant otherwise provides additional information or clarification regarding information already provided
in the submission within the last three months before the goal date.
On
the basis of the FDA’s evaluation of the application and accompanying information, the FDA may issue an approval letter, denial
letter, or a complete response letter. An approval letter authorizes commercial marketing of the product with specific prescribing information
for specific indications. Under the PHSA, the FDA may approve a BLA if it determines that the product is safe, pure and potent and the
facility where the product will be manufactured meets standards designed to ensure that it continues to be safe, pure and potent. If
the application is not approved, the FDA may issue a complete response letter, which will contain the conditions that must be met in
order to secure final approval of the application, and when possible, will outline recommended actions the sponsor might take to obtain
approval of the application. Sponsors that receive a complete response letter may submit to the FDA information that represents a complete
response to the issues identified by the FDA. Such resubmissions are classified under the Prescription Drug User Fee Act, or PDUFA, as
either Class 1 or Class 2, based on the information submitted by an applicant in response to an action letter. Under the goals and policies
agreed to by the FDA under PDUFA, the FDA has two months to review a Class 1 resubmission and six months to review a Class 2 resubmission.
The FDA will not approve an application until issues identified in the complete response letter have been addressed. The FDA issues a
denial letter if it determines that the establishment or product does not meet the agency’s requirements.
The
FDA may also refer the application to an advisory committee for review, evaluation and non-binding recommendation as to whether the application
should be approved. In particular, the FDA may refer applications for novel biologic products or biologic products that present difficult
questions of safety or efficacy to an advisory committee.
If
the FDA approves a new product, the FDA may limit its approved indications for use as well as require that contraindications, warnings
or precautions be included in the product labeling. In addition, the FDA may call for post-approval studies, including Phase 4 clinical
trials, to further assess the product’s safety after approval. The FDA may also require testing and surveillance programs to monitor
the product after commercialization, or impose other conditions, including distribution restrictions or other risk management mechanisms,
including REMS, to help ensure that the benefits of the product outweigh the potential risks. The FDA may prevent or limit further marketing
of a product based on the results of post-market studies or surveillance programs. After approval, many types of changes to the approved
product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements
and FDA review and approval.
Fast
Track, Breakthrough Therapy and Priority Review Designations
The
FDA is authorized to designate certain products for expedited review if they are intended to address an unmet medical need in the treatment
of a serious or life-threatening disease or condition. These programs are referred to as (i) fast track designation, (ii) breakthrough
therapy designation and (iii) priority review designation.
●
Fast Track Review: The FDA may designate a product for fast track review if it is intended (alone or in combination with one or
more other products) for the treatment of a serious or life-threatening disease or condition, and it demonstrates the potential to address
unmet medical needs for such a disease or condition. Sponsors may have greater interactions with the FDA and the FDA may initiate review
of sections of a fast track product’s application before the application is complete. The sponsor must also provide, and the FDA
must approve, a schedule for the submission of the remaining information and the sponsor must pay applicable user fees. However, the
FDA’s time period goal for reviewing a fast track application does not begin until the last section of the application is submitted.
Fast track designation may be withdrawn by the FDA.
●
Breakthrough Therapy: A product may be designated as a breakthrough therapy and be eligible for expedited review if it is intended,
alone or in combination with one or more other products, to treat a serious or life-threatening disease or condition and preliminary
clinical evidence indicates that the product may demonstrate substantial improvement over existing therapies on one or more clinically
significant endpoints, such as substantial treatment effects observed early in clinical development. The FDA may take certain actions
with respect to breakthrough therapies.
●
Priority Review: The FDA may designate a product for priority review if such product treats a serious condition and, if approved,
would provide a significant improvement in safety or effectiveness when compared with other available therapies. This assessment is made
by the FDA on a case-by-case basis. A priority designation is intended to direct overall attention and resources to the evaluation of
such applications, and to shorten the FDA’s goal for taking action on a marketing application from 10 to six months.
Accelerated
Approval Pathway
The
FDA may grant accelerated approval to a product for a serious or life-threatening condition that provides meaningful therapeutic advantage
to patients over existing treatments based upon a determination that the product has an effect on a surrogate endpoint that is reasonably
likely to predict clinical benefit. The FDA may also grant accelerated approval for such a condition when the product has an effect on
an intermediate clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality, or IMM, and that
is reasonably likely to predict an effect on IMM or other clinical benefit, taking into account the severity, rarity, or prevalence of
the condition and the availability or lack of alternative treatments. Products granted accelerated approval must meet the same statutory
standards for safety and effectiveness as those granted traditional approval.
For
the purposes of accelerated approval, a surrogate endpoint is a marker, such as a laboratory measurement, radiographic image, physical
sign, or other measure that is thought to predict clinical benefit but is not itself a measure of clinical benefit. Surrogate endpoints
can often be measured more easily or more rapidly than clinical endpoints. An intermediate clinical endpoint is a measurement of a therapeutic
effect that is considered reasonably likely to predict the clinical benefit of a product, such as an effect on IMM. The accelerated approval
pathway is most often used in settings in which the course of a disease is long, and an extended period of time is required to measure
the intended clinical benefit of a product, even if the effect on the surrogate or intermediate clinical endpoint occurs rapidly. Thus,
accelerated approval has been used extensively in the development and approval of products for treatment of a variety of cancers in which
the goal of therapy is generally to improve survival or decrease morbidity and the duration of the typical disease course requires lengthy
and sometimes large trials to demonstrate a clinical or survival benefit.
The
accelerated approval pathway is usually contingent on a sponsor’s agreement to conduct, in a diligent manner, additional post-approval
confirmatory studies to verify and describe the product’s clinical benefit. As a result, a product candidate approved on this basis
is subject to rigorous post-marketing compliance requirements, including the completion of Phase 4 or post-approval clinical trials to
confirm the effect on the clinical endpoint. Failure to conduct required post-approval studies, or confirm a clinical benefit during
post-marketing studies, would allow the FDA to withdraw the product from the market on an expedited basis. All promotional materials
for product candidates approved under accelerated regulations are subject to prior review by the FDA.
Post-Approval
Regulation
Even
if regulatory approval is granted, a marketed product is subject to continuing comprehensive requirements under federal, state and foreign
laws and regulations, including requirements and restrictions regarding adverse event reporting, recordkeeping, marketing, and compliance
with cGMP. Adverse events reported after approval of a drug can result in additional restrictions on the use of a marketed product or
requirements for additional post-marketing studies or clinical trials.
Maintaining
substantial compliance with applicable federal, state and local statutes and regulations requires the expenditure of substantial time
and financial resources. Rigorous and extensive FDA regulation of biological products continues after approval, particularly with respect
to cGMP requirements. Biological product manufacturers and other entities involved in the manufacture and distribution of approved biological
products are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced
inspections by the FDA and certain state agencies for compliance with cGMP requirements and other laws. We will rely, and expect to continue
to rely, on third parties for the production of clinical and commercial quantities of any products that we may commercialize. Manufacturers
of our products are required to comply with applicable requirements in the cGMP regulations, including quality control and quality assurance
and maintenance of records and documentation. Other post-approval requirements applicable to biological products include record-keeping
requirements, reporting of adverse effects and reporting updated safety and efficacy information.
Discovery
of previously unknown problems or the failure to comply with the applicable regulatory requirements relating to the manufacturer or promotion
of an approved product may result in restrictions on the marketing of a product or withdrawal of the product from the market as well
as significant administrative, civil or criminal sanctions.
Orphan
Drug Designation
Orphan
drug designation in the United States is designed to encourage sponsors to develop products intended for rare diseases or conditions.
In the United States, a rare disease or condition is statutorily defined as a condition that affects fewer than 200,000 individuals in
the United States or that affects more than 200,000 individuals in the United States and for which there is no reasonable expectation
that the cost of developing and making available the product for the disease or condition will be recovered from sales of the product
in the United States.
Orphan
drug designation qualifies a company for tax credits and market exclusivity for seven years following the date of the product’s
marketing approval if granted by the FDA. An application for designation as an orphan product can be made any time prior to the filing
of an application for approval to market the product. A product may be designated as an orphan drug by the FDA Office of Orphan Products
Development, or OOPD, based on an acceptable application. The product must then go through the review and approval process like any other
product. Orphan drug designations may be revoked based on a change in the incidence of the disease.
A
sponsor may request orphan drug designation of a previously unapproved product or a new orphan indication for an already marketed product.
In addition, a sponsor of a product that is otherwise the same product as an already approved orphan drug may seek and obtain orphan
drug designation for the subsequent product for the same rare disease or condition if it can present a plausible hypothesis that its
product may be clinically superior to the first drug. More than one sponsor may receive orphan drug designation for the same product
for the same rare disease or condition, but each sponsor seeking orphan drug designation must file a complete request for designation.
The
period of exclusivity begins on the date that the marketing application is approved by the FDA and applies only to the indication for
which the product has been designated. The FDA may approve a second application for the same product for a different use or a second
application for a clinically superior version of the product for the same use. The FDA cannot, however, approve the same product made
by another manufacturer for the same indication during the market exclusivity period unless it has the consent of the sponsor or the
sponsor is unable to provide sufficient quantities.
Pediatric
Research
Under
the Pediatric Research Equity Act, certain applications for approval must include an assessment, generally based on clinical study data,
of the safety and effectiveness of the subject drug in relevant pediatric populations. The FDA may waive or defer the requirement for
a pediatric assessment, either at the company’s request or by the FDA’s initiative. The FDA may determine that a Risk Evaluation
and Mitigation Strategy are necessary to ensure that the benefits of a new product outweigh its risks. REMS may include various elements,
ranging from a medication guide or patient package insert to limitations on who may prescribe or dispense the drug, depending on what
the FDA considers necessary for the safe use of the drug. Sponsors are required to submit an initial pediatric study plan to their IND
after their end-of-phase 2 meeting with the FDA
Regulation
and Procedures Governing Approval of Medicinal Products in the European Union
In
order to market any product outside of the United States, a company also must comply with numerous regulatory requirements of other countries
and jurisdictions. Whether or not it obtains FDA approval for a product, an applicant will need to obtain the necessary approvals by
the comparable foreign regulatory authorities before it can initiate clinical trials or marketing of the product in those countries or
jurisdictions.
Clinical
Trial Approval
Pursuant
to the currently applicable Clinical Trials Directive 2001/20/EC and the Directive 2005/28/EC on GCP, a system for the approval of clinical
trials in the European Union has been implemented through national legislation of the Member States. Under this system, an applicant
must obtain approval from the competent national authority of a European Union Member State in which the clinical trial is to be conducted
or in multiple Member States if the clinical trial is to be conducted in a number of Member States. Furthermore, the applicant may only
start a clinical trial at a specific study site after the independent ethics committee has issued a favorable opinion. The clinical trial
application, or CTA, must be accompanied by an investigational medicinal product dossier with supporting information prescribed by Directive
2001/20/EC and Directive 2005/28/EC and corresponding national laws of the Member States and further detailed in applicable guidance
documents.
In
April 2014, the European Union adopted a new Clinical Trials Regulation (EU) No 536/2014, which is set to replace the current Clinical
Trials Directive 2001/20/EC. It is expected that the new Clinical Trials Regulation will apply in 2019 or 2020. It will overhaul the
current system of approvals for clinical trials in the European Union. Specifically, the new regulation, which will be directly applicable
in all Member States, aims at simplifying and streamlining the approval of clinical trials in the European Union. For instance, the new
Clinical Trials Regulation provides for a streamlined application procedure using a single entry point and strictly defined deadlines
for the assessment of clinical trial applications.
Marketing
Authorization
To
obtain a marketing authorization for a product under the European Union regulatory system, an applicant must submit an MAA, either under
a centralized procedure administered by the EMA or one of the procedures administered by competent authorities in European Union Member
States (decentralized procedure, national procedure, or mutual recognition procedure). A marketing authorization may be granted only
to an applicant established in the European Union. An applicant must demonstrate compliance with all measures included in an EMA-approved
Pediatric Investigation Plan, or PIP, covering all subsets of the pediatric population, unless the EMA has granted a product-specific
waiver, class waiver, or a deferral for one or more of the measures included in the PIP.
The
centralized procedure provides for the grant of a single marketing authorization by the European Commission that is valid for all European
Union Member States. It is compulsory for specific products, including for medicines produced by certain biotechnological processes,
products designated as orphan medicinal products, advanced therapy products and products with a new active substance indicated for the
treatment of certain diseases, including products for the treatment of cancer. For products with a new active substance indicated for
the treatment of other diseases and products that are highly innovative or for which a centralized process is in the interest of patients,
the centralized procedure may be optional.
Under
the centralized procedure, the Committee for Medicinal Products for Human Use, or the CHMP, established at the EMA is responsible for
conducting the assessment of a product to define its risk/benefit profile. Under the centralized procedure, the maximum timeframe for
the evaluation of an MAA is 210 days, excluding clock stops when additional information or written or oral explanation is to be provided
by the applicant in response to questions of the CHMP. Accelerated evaluation may be granted by the CHMP in exceptional cases, when a
medicinal product is of major interest from the point of view of public health and, in particular, from the viewpoint of therapeutic
innovation.
Periods
of Authorization and Renewals
A
marketing authorization is valid for five years, in principle, and it may be renewed after five years on the basis of a reevaluation
of the risk benefit balance by the EMA or by the competent authority of the authorizing Member State. Once renewed, the marketing authorization
is valid for an unlimited period, unless the European Commission or the competent authority decides, on justified grounds relating to
pharmacovigilance, to proceed with one additional five-year renewal period. Any authorization that is not followed by the placement of
the drug on the European Union market (in the case of the centralized procedure) or on the market of the authorizing Member State within
three years after authorization ceases to be valid.
Regulatory
Requirements after Marketing Authorization
Following
approval, the holder of the marketing authorization is required to comply with a range of requirements applicable to the manufacturing,
marketing, promotion and sale of the medicinal product. These include compliance with the European Union’s stringent pharmacovigilance
or safety reporting rules, pursuant to which post-authorization studies and additional monitoring obligations can be imposed. In addition,
the manufacturing of authorized products, for which a separate manufacturer’s license is mandatory, must also be conducted in strict
compliance with the EMA’s GMP requirements and comparable requirements of other regulatory bodies in the European Union, which
mandate the methods, facilities and controls used in manufacturing, processing and packing of drugs to assure their safety and identity.
Finally, the marketing and promotion of authorized products, including industry-sponsored continuing medical education and advertising
directed toward the prescribers of drugs and/or the general public, are strictly regulated in the European Union under Directive 2001/83EC,
as amended.
Orphan
Drug Designation and Exclusivity
Regulation
(EC) No. 141/2000 and Regulation (EC) No. 847/2000 provide that a product can be designated as an orphan drug by the European Commission
if its sponsor can establish: that the product is intended for the diagnosis, prevention or treatment of (1) a life-threatening or chronically
debilitating condition affecting not more than five in ten thousand persons in the European Union when the application is made, or (2)
a life-threatening, seriously debilitating or serious and chronic condition in the European Union and that without incentives it is unlikely
that the marketing of the drug in the European Union would generate sufficient return to justify the necessary investment. For either
of these conditions, the applicant must demonstrate that there exists no satisfactory method of diagnosis, prevention, or treatment of
the condition in question that has been authorized in the European Union or, if such method exists, the drug has to be of significant
benefit compared to products available for the condition. An orphan drug designation provides benefits such as fee reductions, regulatory
assistance and the possibility to apply for a centralized European Union marketing authorization. Marketing authorization for an orphan
drug leads to a ten-year period of market exclusivity. The market exclusivity period may however be reduced to six years if, at the end
of the fifth year, it is established that the product no longer meets the criteria for orphan drug designation.
Combination
Products in the United States
Certain
products, the combination products, may be comprised of components that would normally be regulated under different types of regulatory
authorities and frequently by different centers at the FDA. A combination product may be (i) a product comprised of two or more regulated
components that are physically, chemically, or otherwise combined or mixed and produced as a single entity; (ii) two or more separate
products packaged together in a single package or as a unit and comprised of drug and device products, device and biological products,
or biological and drug products; (iii) drug, or device, or biological product packaged separately that according to its investigational
plan or proposed labeling is intended for use only with an approved individually specified drug, or device, or biological product where
both are required to achieve the intended use, indication, or effect and where upon approval of the proposed product the labeling of
the approved product would need to be changed, e.g., to reflect a change in intended use, dosage form, strength, route of administration,
or significant change in dose; or (iv) any investigational drug, device, or biological product packaged separately that according to
its proposed labeling is for use only with another individually specified investigational drug, device, or biological product where both
are required to achieve the intended use, indication, or effect. The FDA is charged with assigning a center with primary jurisdiction,
or a lead center, for review of a combination product, this determination being based on the “primary mode of action” of
the combination product. Sponsors may request a jurisdiction determination by submitting a Request for Designation to the office of Combination
Drug Products.
Merger
with Chanticleer and Acquisition of Relief
This
Annual Report on Form 10-K is filed by Sonnet BioTherapeutics Holdings, Inc. (“Sonnet Holdings,” “we,” “us,”
“our,” or the “Company”), formerly known as Chanticleer Holdings, Inc. Until March 31, 2020, the Company was
in the business of owning, operating and franchising fast casual dining concepts domestically and internationally. As previously disclosed,
on April 1, 2020, the Company completed its merger transaction with Sonnet BioTherapeutics, Inc. (“Sonnet”), pursuant to
which Sonnet became a wholly-owned subsidiary of the Company (the “Merger”). On April 1, 2020, in connection with the Merger,
the Company changed its name to “Sonnet BioTherapeutics Holdings, Inc.” Sonnet was incorporated as a New Jersey corporation
on April 6, 2015.
The
Merger was treated by the Company as a reverse merger and accounted for as a reverse recapitalization in accordance with U.S. generally
accepted accounting principles (“U.S. GAAP”). For accounting purposes, Sonnet is considered to have acquired the Company.
In
connection with and prior to the Merger, the Company contributed and transferred to Amergent Hospitality Group, Inc. (“Amergent”),
a newly formed, wholly owned subsidiary of the Company, all of the assets and liabilities relating to the Company’s restaurant
business. The dividend, which together with the contribution and transfer of the Company’s restaurant business described above,
is referred to as the “Spin-Off.” Prior to the Spin-Off, Amergent engaged in no business or operations.
As
a result of the Spin-Off and the Merger, since April 1, 2020, the Company has operated through Sonnet and its direct and indirect subsidiaries
and the ongoing business of the Company is the Sonnet business.
In
addition, in connection with and prior to the Merger, on April 1, 2020, Sonnet completed its acquisition of the global development rights
for Atexakin Alfa (low dose formulation of Interleukin-6, IL-6, now “SON-080”) from Relief Therapeutics Holding SA (“Relief
Holding”) through its acquisition of Relief Holding’s wholly-owned subsidiary, Relief Therapeutics SA (“Relief”),
in exchange for the issuance to Relief Holding of shares of Sonnet common stock that converted into an aggregate of 2,460 shares of Company
common stock in the Merger.
Corporate
and Available Information
The
Company was organized on October 21, 1999, under its original name, Tulvine Systems, Inc., under the laws of the State of Delaware. On
April 25, 2005, Tulvine Systems, Inc. formed a wholly owned subsidiary, Chanticleer Holdings, Inc., and on May 2, 2005, Tulvine Systems,
Inc. merged with, and changed its name to, Chanticleer Holdings, Inc. On April 1, 2020, the Company completed its business combination
with Sonnet BioTherapeutics, Inc. (“Sonnet”), in accordance with the terms of the Agreement and Plan of Merger, dated as
of October 10, 2019, as amended, by and among the Company, Sonnet and Biosub Inc., a wholly-owned subsidiary of the Company (“Merger
Sub”) (the “Merger Agreement”), pursuant to which Merger Sub merged with and into Sonnet, with Sonnet surviving as
a wholly owned subsidiary of the Company (the “Merger”). Under the terms of the Merger Agreement, the Company issued shares
of common stock to Sonnet’s stockholders at an exchange rate of 0.106572 shares for each share of Sonnet common stock outstanding
immediately prior to the Merger. In connection with the Merger, the Company changed its name from “Chanticleer Holdings, Inc.”
to “Sonnet BioTherapeutics Holdings, Inc.,” and the business conducted by the Company became the business conducted by Sonnet.
Our
principal executive offices are located at 100 Overlook Center, Suite 102, Princeton, New Jersey 08540. Our telephone number is (609)
375-2227 and the corporate website address is https://www.sonnetbio.com/. We included the website address in this annual report on Form
10-K only as an inactive textual reference and do not intend it to be an active link to our website. The information on the website is
not incorporated by reference in this annual report on Form 10-K.
This
annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and all amendments to those reports, as well
as other documents we file with the U.S. Securities and Exchange Commission (“SEC”), are available free of charge through
the Investors section of our website as soon as reasonably practicable after such material is electronically filed with or furnished
to the SEC. The public can obtain documents that we file with the SEC at www.sec.gov.
Item
1A. Risk Factors.
An
investment in our common stock involves a high degree of risk including the risk of a loss of your entire investment. You should carefully
consider the risks and uncertainties described below and the other information contained in this report and the other reports filed by
us with the SEC. The risks set forth below are not the only ones facing us. Additional risks and uncertainties may exist that could also
adversely affect our business, operations and financial condition. If any of the following risks actually materialize, our business,
financial condition and/or operations could suffer. In such event, the value of our common stock could decline, and you could lose all
or a substantial portion of the money that you pay for our common stock.
Summary
of Risk Factors
| |
● |
We
have a history of significant operating losses and expect to incur significant and increasing losses for the foreseeable future,
and we may never achieve or maintain profitability. |
| |
● |
Our
recurring losses from operations have raised substantial doubt regarding our ability to continue as a going concern. |
| |
● |
We
will need significant additional capital, and if we are unable to raise capital when needed, we could be forced to delay, reduce
or eliminate our product discovery and development programs or commercialization efforts. |
| |
● |
We
are substantially dependent on the success of our internal development programs and our product pipeline candidates may not successfully
complete clinical trials, receive regulatory approval or be successfully commercialized. |
| |
● |
We
are at a very early stage in our development efforts, our product candidates represent a new category of medicines and may be subject
to heightened regulatory scrutiny until they are established as a therapeutic modality. |
| |
● |
Even
if we complete the necessary preclinical studies and clinical trials, the marketing approval process is expensive, time consuming
and uncertain and may prevent us or any collaborators from obtaining approvals for the commercialization of some or all of our product
candidates. As a result, we cannot predict when or if, and in which territories, we, or any collaborators, will obtain marketing
approval to commercialize a product candidate. |
| |
● |
We
face significant competition and if our competitors develop and market products that are more effective, safer or less expensive
than the product candidates we develop, our commercial opportunities will be negatively impacted. |
| |
● |
The
commercial success of any current or future product candidate will depend upon the degree of market acceptance by physicians, patients,
payors and others in the medical community. |
| |
● |
For
certain product candidates, we may depend on development and commercialization collaborators to develop and conduct clinical trials
with, obtain regulatory approvals for, and if approved, market and sell product candidates. If such collaborators fail to perform
as expected, the potential for us to generate future revenue from such product candidates would be significantly reduced and our
business would be harmed. |
| |
● |
We
will rely on third parties, including independent clinical investigators and CROs, to conduct and sponsor some of the clinical trials
of our product candidates. Any failure by a third party to meet its obligations with respect to the clinical development of our product
candidates may delay or impair our ability to obtain regulatory approval for our product candidates. |
| |
● |
If
we are unable to obtain and maintain patent and other intellectual property protection for our products and product candidates, or
if the scope of the patent and other intellectual property protection obtained is not sufficiently broad, our competitors could develop
and commercialize products similar or identical to ours, and our ability to successfully commercialize our products and product candidates
may be adversely affected. |
| |
● |
We
expect to expand our organization, and as a result, we may encounter difficulties in managing our growth, which could disrupt our
operations. |
| |
● |
We
do not expect to pay cash dividends in the foreseeable future and therefore investors should not anticipate cash dividends on their
investment. |
Risks
Related to Our Financial Position and Need for Additional Capital
We
have a history of significant operating losses and expect to incur significant and increasing losses for the foreseeable future, and
we may never achieve or maintain profitability.
We
do not expect to generate revenue or profitability that is necessary to finance our operations in the short term. Our net losses for
the years ended September 30, 2023 and 2022 were $18.8 million and $29.7 million, respectively. As of September 30, 2023, we had an accumulated
deficit of $110.2 million.
To
date, we have not commercialized any products or generated any revenues from the sale of products, and absent the realization of sufficient
revenues from product sales, we may never attain profitability in the future. We have devoted substantially all of our financial resources
and efforts to research and development, including preclinical studies and our clinical trials. Our net losses may fluctuate significantly
from quarter to quarter and year to year. Net losses and negative cash flows have had, and will continue to have, an adverse effect on
our shareholders’ (deficit) equity and working capital.
We
anticipate that our expenses will increase substantially if and as we:
●
continue to develop and conduct clinical trials with respect to our lead product candidate, SON-080, and our other product candidates;
●
initiate and continue research, preclinical and clinical development efforts for any future product candidates;
●
seek to discover and develop additional product candidates and further expand our clinical product pipeline;
●
seek marketing and regulatory approvals for any product candidates that successfully complete clinical trials;
●
require the manufacture of larger quantities of product candidates for clinical development and, potentially, commercialization;
●
maintain, expand and protect our intellectual property portfolio;
●
expand our research and development infrastructure, including hiring and retaining additional personnel, such as clinical, quality control
and scientific personnel;
●
establish sales, marketing, distribution and other commercial infrastructure in the future to commercialize products for which we obtain
marketing approval, if any;
●
add operational, financial and management information systems and personnel, including personnel to support our product development and
commercialization and help us comply with our obligations as a public company; and
●
add equipment and physical infrastructure to support our research and development.
Our
ability to become and remain profitable depends on our ability to license our products and generate revenue. Generating product revenue
will depend on our ability to obtain marketing approval for, and successfully commercialize, one or more of our product candidates.
Successful
commercialization will require achievement of key milestones, including completing clinical trials of our product candidates, obtaining
marketing approval for these product candidates, manufacturing, marketing and selling those products for which we, or any collaborators,
may obtain marketing approval, satisfying any post-marketing requirements and obtaining reimbursement for our products from private insurance
or government payors. Because of the uncertainties and risks associated with these activities, we are unable to accurately predict the
timing and amount of revenues, and if or when we might achieve profitability. We and any collaborators may never succeed in these activities
and, even if we do, or any collaborators do, we may never generate revenues that are large enough for us to achieve profitability. Even
if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis.
Our
failure to become and remain profitable would depress the market price of our common stock and could impair our ability to raise capital,
expand our business, diversify our product offerings or continue our operations. If we continue to suffer losses, investors may not receive
any return on their investment and may lose their entire investment.
Our
limited operating history may make it difficult for you to evaluate the success of our business to date and to assess our future viability.
Our
business commenced operations in 2015. Our operations to date have been limited to financing and staffing our company, developing our
technology, conducting preclinical research and early-stage clinical trials for our product candidates and pursuing strategic collaborations
to advance our product candidates. We have not yet demonstrated an ability to successfully conduct late-stage clinical trials, obtain
marketing approvals, manufacture a commercial-scale product, or arrange for a third party to do so on our behalf, or conduct sales and
marketing activities necessary for successful product commercialization. Accordingly, you should consider our prospects in light of the
costs, uncertainties, delays and difficulties frequently encountered by companies in the early stages of development, especially clinical-stage
biopharmaceutical companies such as ours. Any predictions you make about our future success or viability may not be as accurate as they
would be if we had a longer operating history or a history of successfully developing and commercializing pharmaceutical products.
We
may encounter unforeseen expenses, difficulties, complications, delays and other known or unknown factors in achieving our business objectives.
We will eventually need to transition from a company with a development focus to a company capable of supporting commercial activities.
We may not be successful in such a transition.
We
expect our financial condition and operating results to continue to fluctuate significantly from quarter to quarter and year to year
due to a variety of factors, many of which are beyond our control. Accordingly, you should not rely upon the results of any quarterly
or annual periods as indications of future operating performance.
Our
recurring losses from operations have raised substantial doubt regarding our ability to continue as a going concern.
We
have incurred recurring losses and negative cash flows from operations activities since inception and we expect to generate losses and
negative cash flows from operations for the foreseeable future primarily due to research and development costs for our potential product
candidates. As of September 30, 2023, we had cash of $2.3 million and stockholders’ deficit of $0.2 million. We believe our cash
at September 30, 2023, together with the $4.1 million in net proceeds received from our public offering of common stock and warrants
on October 24, 2023, will fund our projected operations into March 2024. We also expect to receive
a $0.8 million net cash refund from the research and development tax incentive program in Australia (see Note 2 to the consolidated financial
statements) and recently received preliminary approval of our application to sell up to $4.8 million of our New Jersey state net operating
losses through the Technology Business Tax Certificate Transfer Program, subject to execution of such sale (see Note 10 to the consolidated
financial statements). Substantial additional financing will be needed by
us to fund our operations. These factors raise substantial doubt about our ability to continue as a going concern. The accompanying consolidated
financial statements have been prepared on a going concern basis, which contemplates the realization of assets and satisfaction of liabilities
in the normal course of business. The consolidated financial statements do not include any adjustments that might result from the outcome
of this uncertainty.
We
will require additional capital in the future through equity or debt financings, partnerships, collaborations, or other sources to carry
out our planned development activities. If additional capital is not secured when required, we may need to delay or curtail our operations
until such funding is received. Various internal and external factors will affect whether and when our product candidates become approved
for marketing and successful commercialization. The regulatory approval and market acceptance of our products candidates, length of time
and cost of developing and commercializing these product candidates and/or failure of them at any stage of the approval process will
materially affect our financial condition and future operations.
Operations
since inception have consisted primarily of organizing us, securing financing, developing its technologies through performing research
and development and conducting preclinical studies. We face risks associated with companies whose products are in development. These
risks include the need for additional financing to complete its research and development, achieving its research and development objectives,
defending its intellectual property rights, recruiting and retaining skilled personnel, and dependence on key members of management.
Our
ability to continue as a going concern is dependent on our ability to raise additional equity or debt capital or spin-off non-core assets
to raise additional cash. Should we be unable to raise sufficient additional capital, we may be required to undertake cost-cutting measures
including delaying or discontinuing certain clinical activities.
The
source, timing and availability of any future financing will depend principally upon market conditions, and, more specifically, on the
progress of our clinical development programs. Funding may not be available when needed, at all, or on terms acceptable to us. Lack of
necessary funds may require us, among other things, to delay, scale back or eliminate some or all of our planned clinical trials. These
factors among others create a substantial doubt about our ability to continue as a going concern.
We
will need substantial additional funding, and if we are unable to raise capital when needed, we could be forced to delay, reduce or eliminate
our product discovery and development programs or commercialization efforts.
Developing
pharmaceutical products, including conducting preclinical studies and clinical trials, is a very time-consuming, expensive and uncertain
process that takes years to complete. For example, for the years ended September 30, 2023 and 2022, we used $21.3 million and $27.8 million,
respectively, in net cash for our operating activities, substantially all of which related to research and development activities. We
expect our expenses to increase in connection with our ongoing activities, particularly as we initiate new clinical trials of, initiate
new research and preclinical development efforts for and seek marketing approval for, our current product candidates or any future product
candidates. In addition, if we obtain marketing approval for any of our product candidates, we may incur significant commercialization
expenses related to product sales, marketing, manufacturing and distribution to the extent that such sales, marketing, manufacturing
and distribution are not the responsibility of a collaborator. Furthermore, as a result of the Merger, we will continue to incur significant
costs associated with operating as a public company. Accordingly, we will need to obtain substantial additional funding in connection
with our continuing operations. If we are unable to raise capital when needed or on attractive terms, we may be forced to delay, reduce
or eliminate our research and development programs or any future commercialization efforts.
We
will be required to expend significant funds in order to advance the development of the product candidates in our pipeline, as well as
other product candidates we may seek to develop. In addition, while we may seek one or more collaborators for future development of our
product candidates, we may not be able to enter into a collaboration for any of our product candidates for such indications on suitable
terms, on a timely basis or at all. In any event, our existing cash will not be sufficient to fund all of the efforts that we plan to
undertake or to fund the completion of development of any of our product candidates. Accordingly, we will be required to obtain further
funding through public or private equity offerings, debt financings, collaborations and licensing arrangements or other sources. We do
not have any committed external source of funds. Adequate additional financing may not be available to us on acceptable terms, or at
all. Our failure to raise capital as and when needed would have a negative impact on our financial condition and our ability to pursue
our business strategy.
Our
estimate may prove to be wrong, and we could use our available capital resources sooner than we currently expect. Further, changing circumstances,
some of which may be beyond our control, could cause us to consume capital significantly faster than we currently anticipate, and we
may need to seek additional funds sooner than planned. Our future funding requirements, both short-term and long-term, will depend on
many factors, including:
●
the scope, progress, timing, costs and results of clinical trials of, and research and preclinical development efforts for, our current
and future product candidates;
●
our ability to enter into, and the terms and timing of, any collaborations, licensing or other arrangements;
●
our ability to identify one or more future product candidates for our pipeline;
●
the number of future product candidates that we pursue and their development requirements;
●
the outcome, timing and costs of seeking regulatory approvals;
●
the costs of commercialization activities for any of our product candidates that receive marketing approval to the extent such costs
are not the responsibility of any collaborators, including the costs and timing of establishing product sales, marketing, distribution
and manufacturing capabilities;
●
the receipt of marketing approval, revenue, if any, received from commercial sales of our current and future product candidates;
●
our headcount growth and associated costs as we expand our research and development and establish a commercial infrastructure;
●
the costs of preparing, filing and prosecuting patent applications, maintaining and protecting our intellectual property rights including
enforcing and defending intellectual property related claims; and
●
the costs of operating as a public company.
Raising
additional capital may cause dilution to our existing shareholders, restrict our operations or cause us to relinquish valuable rights.
We
may seek additional capital through a combination of public and private equity offerings, debt financings, strategic partnerships and
alliances, licensing arrangements or monetization transactions. To the extent that we raise additional capital through the sale of equity,
convertible debt securities or other equity-based derivative securities, your ownership interest will be diluted and the terms may include
liquidation or other preferences that adversely affect your rights as a shareholder. Any indebtedness we incur would result in increased
fixed payment obligations and could involve restrictive covenants, such as limitations on our ability to incur additional debt, limitations
on our ability to acquire or license intellectual property rights and other operating restrictions that could adversely impact our ability
to conduct our business. Furthermore, the issuance of additional securities, whether equity or debt, by us, or the possibility of such
issuance, may cause the market price of our common stock to decline and existing shareholders may not agree with our financing plans
or the terms of such financings. If we raise additional funds through strategic partnerships and alliances, licensing arrangements or
monetization transactions with third parties, we may have to relinquish valuable rights to our technologies, or our product candidates,
or grant licenses on terms unfavorable to us. Adequate additional financing may not be available to us on acceptable terms, or at all.
If we are unable to raise additional funds when needed, we may be required to delay, limit, reduce or terminate our product development
or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop
and market ourselves.
Risks
Related to the Discovery, Development and Regulatory Approval of Our Product Candidates
The
coronavirus COVID-19 pandemic or the widespread outbreak of any other communicable disease could materially and adversely affect our
business, financial condition and results of operations.
We
face risks related to health epidemics or outbreaks of communicable diseases, for example, the recent outbreak around the world of the
highly transmissible and pathogenic coronavirus COVID-19. The outbreak of such communicable diseases could result in a widespread health
crisis that could adversely affect general commercial activity and the economies and financial markets of many countries.
In
December 2019, a novel strain of coronavirus, COVID-19, was reported to have surfaced in Wuhan, China and on March 11, 2020 was declared
a pandemic by the World Health Organization. The extent to which COVID-19 may impact our preclinical and clinical trial operations will
depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the duration and geographic
reach of the outbreak, the severity of COVID-19, and the effectiveness of actions to contain and treat COVID-19.
Many
countries around the world imposed quarantines and restrictions on travel and mass gatherings to slow the spread of COVID-19 and have
closed non-essential businesses, though such restrictions have currently lapsed in most cases. Such events may result in a period of
business, supply and drug product manufacturing disruption, and in reduced operations, any of which could materially affect our business,
financial condition and results of operations.
This
pandemic or outbreak could result in difficulty securing clinical trial site locations, CROs, and/or trial monitors and other critical
vendors and consultants supporting the trial. In addition, outbreaks or the perception of an outbreak near a clinical trial site location
could impact our ability to enroll patients. These situations, or others associated with COVID-19, could cause delays in our clinical
trial plans and could increase expected costs, all of which could have a material adverse effect on our business and its financial condition.
In
particular, manufacturing of our pipeline products (other than SON-1010) was delayed by COVID-19 related supply chain issues, specifically
supply of raw materials, including media, resins, and analytical kits, compounded by international shipping delays. However, COVID-19
is not currently impacting our business.
While
the potential economic impact brought by, and the duration of, COVID-19 or the widespread outbreak of any other communicable disease
may be difficult to assess or predict, a widespread pandemic could result in significant disruption of global financial markets,
reducing our ability to access capital, which could in the future negatively affect our liquidity. In addition, a recession or
market correction resulting from the spread of COVID-19 or any other communicable disease could materially affect our business and
the value of our common shares.
An outbreak may also affect the ability of our staff and the parties we work with to carry out our non-clinical, clinical, and
drug manufacturing activities. We rely or may in the future rely on clinical sites, investigators and other study staff, consultants,
independent contractors, contract research organizations and other third-party service providers to assist us in managing, monitoring
and otherwise carrying out our nonclinical studies and clinical trials. We also rely or may in the future rely on consultants, independent
contractors, contract manufacturing organizations, and other third-party service providers to assist us in managing, monitoring and otherwise
carrying out our API production, formulation, and drug manufacturing activities. A widespread pandemic would affect the ability of any of these external
people, organizations, or companies to devote sufficient time and resources to our programs or to travel to perform work for us.
Potential
negative impacts of the COVID-19 outbreak or the widespread outbreak of any other communicable disease on the conduct of current or
future clinical studies include delays in gaining feedback from regulatory agencies, starting new clinical studies, and recruiting
subjects to studies that are enrolling. The potential negative impacts also include inability to have study visits at study sites,
incomplete collection of safety and efficacy data, and higher rates of drop-out of subjects from ongoing studies, delays in site
entry of study data into the data base, delays in monitoring of study data because of restricted physical access to study sites,
delays in site responses to queries, delays in data-base lock, delays in data analyses, delays in time to top-line data, and delays
in completing study reports. New or worsening COVID-19 (or any other communicable disease) disruptions or restrictions could have
the potential to further negatively impact our non-clinical studies, clinical trials, and drug manufacturing activities.
We
are substantially dependent on the success of our internal development programs and our product pipeline candidates may not successfully
complete clinical trials, receive regulatory approval or be successfully commercialized.
Our
future success will depend heavily on the success of our internal development programs and of product candidates from our pipeline program.
Our
ability to successfully commercialize our pipeline and our other product candidates will depend on, among other things, our ability to:
●
successfully complete preclinical studies and clinical trials;
●
receive regulatory approvals from the FDA, the EMA and other similar regulatory authorities;
●
establish and maintain collaborations with third parties for the development and/or commercialization of our product candidates, or otherwise
build and maintain strong development, sales, distribution and marketing capabilities that are sufficient to develop products and launch
commercial sales of any approved products;
●
obtain coverage and adequate reimbursement from payors such as government health care systems and insurance companies and achieve commercially
attractive levels of pricing;
●
secure acceptance of our product candidates from physicians, health care payors, patients and the medical community;
●
produce, through a validated process, in manufacturing facilities inspected and approved by regulatory authorities, including the FDA,
sufficiently large quantities of our product candidates to permit successful commercialization;
●
manage our spending as expenses increase due to clinical trials and commercialization; and
●
obtain and enforce sufficient intellectual property rights for any approved products and product candidates.
Of
the large number of drugs in development in the pharmaceutical industry, only a small percentage result in the submission of a new drug
application, or NDA, or biologics licensing application, or BLA, to the FDA and even fewer are approved for commercialization. Furthermore,
even if we do receive regulatory approval to market our product candidates, any such approval may be subject to limitations on the indicated
uses or patient populations for which we may market the product. Accordingly, even if we are able to obtain the requisite financing to
continue to fund our development programs, we cannot assure you that our product candidates will be successfully developed or commercialized.
If we are unable to develop, or obtain regulatory approval for, or, if approved, to successfully commercialize our product candidates,
we may not be able to generate sufficient revenue to continue our business.
We
are at a very early stage in our development efforts, our product candidates represent a new category of medicines and may be subject
to heightened regulatory scrutiny until they are established as a therapeutic modality.
Our
pipeline product candidates represent a new therapeutic modality of including engaging a Fully Human Albumin Binding Domain to deliver
therapeutic products. Our product candidates may not demonstrate in patients any or all of the pharmacological benefits we believe they
may possess. We have not yet succeeded and may never succeed in demonstrating efficacy and safety for these or any other product candidates
in clinical trials or in obtaining marketing approval thereafter.
Regulatory
authorities do not have experience with our product candidate and may require evidence of safety and efficacy that goes beyond what we
have included in our development plans. In such a case, development of our product candidates may be more costly or time-consuming than
expected, and our candidate products may not prove to be viable.
If
we are unsuccessful in our development efforts, we may not be able to advance the development of our product candidates, commercialize
products, raise capital, expand our business or continue our operations.
Our
product candidates and those of any collaborators will need to undergo preclinical and clinical trials that are time-consuming and expensive,
the outcomes of which are unpredictable, and for which there is a high risk of failure. If preclinical or clinical trials of our or their
product candidates fail to satisfactorily demonstrate safety and efficacy to the FDA, the EMA and any other comparable regulatory authority,
additional costs may be incurred or delays experienced in completing, the development of these product candidates, or their development
may be abandoned.
The
FDA in the United States, the EMA in the European Union and the European Economic Area, and other comparable regulatory authorities in
other jurisdictions must approve new product candidates before they can be marketed, promoted or sold in those territories. We have not
previously submitted an IND or BLA to the FDA or similar drug approval filings to comparable foreign regulatory authorities for any of
our product candidates. We must provide these regulatory authorities with data from preclinical studies and clinical trials that demonstrate
that our product candidates are safe and effective for a specific indication before they can be approved for commercial distribution.
We cannot be certain that our clinical trials for our product candidates will be successful or that any of our product candidates will
receive approval from the FDA, the EMA or any other comparable regulatory authority.
Preclinical
studies and clinical trials are long, expensive and unpredictable processes that can be subject to extensive delays. We cannot guarantee
that any clinical trials will be conducted as planned or completed on schedule, if at all. It may take several years and require significant
expenditures to complete the preclinical studies and clinical trials necessary to commercialize a product candidate, and delays or failure
are inherently unpredictable and can occur at any stage. We may also be required to conduct additional clinical trials or other testing
of our product candidates beyond the trials and testing that we contemplate, which may lead to us incurring additional unplanned costs
or result in delays in clinical development. In addition, we may be required to redesign or otherwise modify our plans with respect to
an ongoing or planned clinical trial, and changing the design of a clinical trial can be expensive and time consuming. An unfavorable
outcome in one or more trials would be a major setback for our product candidates and for us. An unfavorable outcome in one or more trials
may require us to delay, reduce the scope of or eliminate one or more product development programs, which could have a material adverse
effect on our business, financial position, results of operations and future growth prospects.
Many
of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial
of marketing approval for our product candidates. The FDA, EMA or any other comparable regulatory authority may disagree with our clinical
trial design and our interpretation of data from clinical trials, or may change the requirements for approval even after it has reviewed
and commented on the design for our clinical trials.
In
connection with clinical trials of our product candidates, we face a number of risks, including risks that:
●
a product candidate is ineffective or inferior to existing approved products for the same indications;
●
a product candidate causes or is associated with unacceptable toxicity or has unacceptable side effects;
●
patients may die or suffer adverse effects for reasons that may or may not be related to the product candidate being tested;
●
the results may not confirm the positive results of earlier trials;
●
the results may not meet the level of statistical significance required by the FDA, the EMA or other relevant regulatory agencies to
establish the safety and efficacy of our product candidates for continued trial or marketing approval; and
●
our collaborators may be unable or unwilling to perform under their contracts.
Furthermore,
we sometimes estimate for planning purposes the timing of the accomplishment of various scientific, clinical, regulatory and other product
development objectives. These milestones may include our expectations regarding the commencement or completion of scientific studies,
clinical trials, the submission of regulatory filings or commercialization objectives. From time to time, we may publicly announce the
expected timing of some of these milestones, such as the completion of an ongoing clinical trial, the initiation of other clinical programs,
the receipt of marketing approval or a commercial launch of a product. The achievement of many of these milestones may be outside of
our control. All of these milestones are based on a variety of assumptions, which may cause the timing of achievement of the milestones
to vary considerably from our estimates. If we fail to achieve milestones in the timeframes we expect, the commercialization of our product
candidates may be delayed, we may not be entitled to receive certain contractual payments, which could have a material adverse effect
on our business, financial position, results of operations and future growth prospects.
We
may find it difficult to enroll patients in our clinical trials, which could delay or prevent us from proceeding with clinical trials
of our product candidates.
Identifying
and qualifying patients to participate in clinical trials of our product candidates is critical to our success. The timing of our clinical
trials depends on our ability to recruit patients to participate as well as the completion of required follow-up periods. Patients may
be unwilling to participate in our clinical trials because of negative publicity from adverse events related to novel therapeutic approaches,
competitive clinical trials for similar patient populations, the existence of current treatments or for other reasons. Enrollment risks
are heightened with respect to certain indications that we may target for one or more of our product candidates that may be rare diseases,
which may limit the pool of patients that may be enrolled in our planned clinical trials. The timeline for recruiting patients, conducting
trials and obtaining regulatory approval of our product candidates may be delayed, which could result in increased costs, delays in advancing
our product candidates, delays in testing the effectiveness of our product candidates or termination of the clinical trials altogether.
We
may not be able to identify, recruit and enroll a sufficient number of patients, or those with the required or desired characteristics,
to complete our clinical trials in a timely manner. For example, due to the nature of the indications that we are initially targeting,
patients with advanced disease progression may not be suitable candidates for treatment with our product candidates and may be ineligible
for enrollment in our clinical trials. Therefore, early diagnosis in patients with our target diseases is critical to our success. Patient
enrollment and trial completion is affected by factors including the:
●
size of the patient population and process for identifying subjects;
●
design of the trial protocol;
●
eligibility and exclusion criteria;
●
safety profile, to date, of the product candidate under study;
●
perceived risks and benefits of the product candidate under study;
●
perceived risks and benefits of our approach to treatment of diseases;
●
availability of competing therapies and clinical trials;
●
severity of the disease under investigation;
●
degree of progression of the subject’s disease at the time of enrollment;
●
proximity and availability of clinical trial sites for prospective subjects;
●
ability to obtain and maintain subject consent;
●
risk that enrolled subjects will drop out before completion of the trial;
●
patient referral practices of physicians; and
●
ability to monitor subjects adequately during and after treatment.
In
addition, clinical development for pilot scale feasibility study of SON-080 is currently planned to take place outside of the U.S. Our
ability to successfully initiate, enroll and complete a clinical trial in any foreign country is subject to numerous risks unique to
conducting business in foreign countries, including:
●
difficulty in establishing or managing relationships with academic partners or contract research organizations, or CROs, and physicians;
●
different standards for the conduct of clinical trials;
●
the absence in some countries of established groups with sufficient regulatory expertise for review of protocols related to our novel
approach;
●
our inability to locate qualified local consultants, physicians and partners; and
●
the potential burden of complying with a variety of foreign laws, medical standards and regulatory requirements, including the regulation
of pharmaceutical and biotechnology products and treatment.
If
we have difficulty enrolling a sufficient number of patients to conduct our clinical trials as planned, we may need to delay, limit or
terminate ongoing or planned clinical trials, any of which would have an adverse effect on our business, financial condition, results
of operations and prospects.
Results
of preclinical studies and early clinical trials may not be predictive of results of future clinical trials.
The
outcome of preclinical studies and early clinical trials may not be predictive of the success of later clinical trials, and interim results
of clinical trials do not necessarily predict success in the results of completed clinical trials. Many companies in the pharmaceutical
and biotechnology industries have suffered significant setbacks in late-stage clinical trials after achieving positive results in earlier
development, and we could face similar setbacks. For example, the Phase IIa trial of SON-080 will be conducted outside of the U.S., and
the findings may not be replicated in future trials at global clinical trial sites in a later stage clinical trial conducted by us or
our collaborators. The design of a clinical trial can determine whether its results will support approval of a product and flaws in the
design of a clinical trial may not become apparent until the clinical trial is well advanced. We may be unable to design and execute
a clinical trial to support marketing approval.
Preclinical
and clinical data are often susceptible to varying interpretations and analyses. Many companies that believed their product candidates
performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval for the product
candidates. Even if we, or any collaborators, believe that the results of clinical trials for our product candidates warrant marketing
approval, the FDA or comparable foreign regulatory authorities may disagree and may not grant marketing approval of our product candidates.
In
some instances, there can be significant variability in safety or efficacy results between different clinical trials of the same product
candidate due to numerous factors, including changes in trial procedures set forth in protocols, differences in the size and type of
the patient populations, changes in and adherence to the dosing regimen and other clinical trial protocols and the rate of dropout among
clinical trial participants. If we fail to receive positive results in clinical trials of our product candidates, the development timeline
and regulatory approval and commercialization prospects for our most advanced product candidates, and, correspondingly, our business
and financial prospects would be negatively impacted.
Our
current or future product candidates may cause undesirable side effects or have other properties when used alone or in combination with
other approved products or investigational new drugs that could halt their clinical development, prevent their marketing approval, limit
their commercial potential or result in significant negative consequences.
Undesirable
or clinically unmanageable side effects could occur and cause us or regulatory authorities to interrupt, delay or halt clinical trials
and could result in a more restrictive label or the delay or denial of marketing approval by the FDA or comparable foreign regulatory
authorities. Results of our trials could reveal a high and unacceptable severity and prevalence of side effects or unexpected characteristics.
If
unacceptable side effects arise in the development of our product candidates, we, the FDA or comparable foreign regulatory authorities,
the Institutional Review Boards, or IRBs, or independent ethics committees at the institutions in which our studies are conducted, or
the Data Safety Monitoring Board, or DSMB, could suspend or terminate our clinical trials or the FDA or comparable foreign regulatory
authorities could order us to cease clinical trials or deny approval of our product candidates for any or all targeted indications. Treatment-related
side effects could also affect patient recruitment or the ability of enrolled subjects to complete the trial, or result in potential
product liability claims. In addition, these side effects may not be appropriately recognized or managed by the treating medical staff.
We may be required to train medical personnel using our product candidates to understand the side effect profiles for our clinical trials
and upon any commercialization of any of our product candidates. Inadequate training in recognizing or managing the potential side effects
of our product candidates could result in patient injury or death. Any of these occurrences may prevent us from achieving or maintaining
market acceptance of the affected product candidate and may harm our business, financial condition and prospects significantly.
Moreover,
clinical trials of our product candidates are conducted in carefully defined sets of patients who have agreed to enter into clinical
trials. Consequently, it is possible that our clinical trials may indicate an apparent positive effect of a product candidate that is
greater than the actual positive effect, if any, or alternatively fail to identify undesirable side effects. If, following approval of
a product candidate, we, or others, discover that the product is less effective than previously believed or causes undesirable side effects
that were not previously identified, any of the following consequences could occur:
●
regulatory authorities may withdraw their approval of the product or seize the product;
●
we, or any collaborators, may need to recall the product, or be required to change the way the product is administered or conduct additional
clinical trials;
●
additional restrictions may be imposed on the marketing of, or the manufacturing processes for, the particular product;
●
we may be subject to fines, injunctions or the imposition of civil or criminal penalties;
●
regulatory authorities may require the addition of labeling statements, such as a boxed warning or a contraindication;
●
we, or any collaborators, may be required to create a medication guide outlining the risks of the previously unidentified side effects
for distribution to patients;
●
we, or any collaborators, could be sued and held liable for harm caused to patients;
●
the product may become less competitive; and
●
our reputation may suffer.
If
any of our current or future product candidates fail to demonstrate safety and efficacy in clinical trials or do not gain marketing approval,
we will not be able to generate revenue and our business will be harmed. Any of these events could harm our business and operations,
and could negatively impact the price of our common stock.
We
may not be successful in our efforts to identify or discover additional product candidates.
Although
we intend to explore other therapeutic opportunities in addition to the product candidates that we are currently developing, we may fail
to identify other product candidates for clinical development for a number of reasons. For example, our research methodology may not
be successful in identifying potential product candidates or those we identify may be shown to have harmful side effects or other characteristics
that make them unmarketable or unlikely to receive regulatory approval. Additional product candidates will require additional, time-consuming
development efforts prior to commercial sale, including preclinical studies, clinical trials and approval by the FDA and/or applicable
foreign regulatory authorities. All product candidates are prone to the risks of failure that are inherent in pharmaceutical product
development. If we fail to identify and develop additional potential product candidates, we may be unable to grow our business and our
results of operations could be materially harmed.
We
may expend our limited resources to pursue a particular product candidate or indication and fail to capitalize on product candidates
or indications that may be more profitable or for which there is a greater likelihood of success.
Because
we have limited financial and managerial resources, we intend to focus on developing product candidates for specific indications that
we identify as most likely to succeed, in terms of both their potential for marketing approval and commercialization. As a result, we
may forego or delay pursuit of opportunities with other product candidates or for other indications that may prove to have greater commercial
potential.
Our
resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our
spending on current and future research and development programs and product candidates for specific indications may not yield any commercially
viable product candidates. If we do not accurately evaluate the commercial potential or target market for a particular product candidate,
we may relinquish valuable rights to that product candidate through collaboration, licensing or other royalty arrangements in cases in
which it would have been more advantageous for us to retain sole development and commercialization rights to the product candidate.
We
face potential product liability, and, if successful claims are brought against us, we may incur substantial liability and costs. If
the use of our product candidates harms patients, or is perceived to harm patients even when such harm is unrelated to our product candidates,
our regulatory approvals could be revoked or otherwise negatively impacted and we could be subject to costly and damaging product liability
claims.
The
use of our product candidates in clinical trials and the sale of any products for which we obtain marketing approval expose us to the
risk of product liability claims. Product liability claims might be brought against us by patients, healthcare providers, pharmaceutical
companies or others selling or otherwise coming into contact with our products. There is a risk that our product candidates may induce
adverse events. If we cannot successfully defend against product liability claims, we could incur substantial liability and costs. In
addition, regardless of merit or eventual outcome, product liability claims may result in:
| |
● |
the
impairment of our business reputation; |
| |
|
|
| |
● |
the
withdrawal of clinical trial participants; |
| |
|
|
| |
● |
substantial
monetary awards to patients or other claimants; |
| |
|
|
| |
● |
costs
due to related litigation; |
| |
|
|
| |
● |
the
distraction of management’s attention from our primary business; |
| |
|
|
| |
● |
the
inability to commercialize our product candidates; and |
| |
|
|
| |
● |
decreased
demand for our product candidates, if approved for commercial sale. |
We
intend to acquire product liability insurance coverage in light of our current clinical programs; however, we may not be able to maintain
insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. We intend to expand our
insurance coverage each time we commercialize an additional product; however, we may be unable to obtain product liability insurance
on commercially reasonable terms or in adequate amounts. On occasion, large judgments have been awarded in class action lawsuits based
on drugs or medical treatments that had unanticipated adverse effects. A successful product liability claim or series of claims brought
against us could cause our stock price to decline and, if judgments exceed our insurance coverage, could adversely affect our results
of operations and business.
Patients
with the diseases targeted by certain of our product candidates, such as our lead indications in oncology, are often already in severe
and advanced stages of disease and have both known and unknown significant pre-existing and potentially life-threatening health risks.
During the course of treatment, patients may suffer adverse events, including death, for reasons that may be related to our product candidates.
Such events could subject us to costly litigation, require us to pay substantial amounts of money to injured patients, delay, negatively
impact or end our opportunity to receive or maintain regulatory approval to market our products, or require us to suspend or abandon
our commercialization efforts. Even in a circumstance in which we do not believe that an adverse event is related to our products, the
investigation into the circumstance may be time-consuming or inconclusive. These investigations may interrupt our sales efforts, delay
our regulatory approval process, or impact and limit the type of regulatory approvals our product candidates receive or maintain. As
a result of these factors, a product liability claim, even if successfully defended, could have a material adverse effect on our business,
financial condition or results of operations.
We
may seek designations for our product candidates with the FDA and other comparable regulatory authorities that are intended to confer
benefits such as a faster development process or an accelerated regulatory pathway, but there can be no assurance that we will successfully
obtain such designations. In addition, even if one or more of our product candidates are granted such designations, we may not be able
to realize the intended benefits of such designations.
The
FDA and other comparable regulatory authorities offer certain designations for product candidates that are intended to encourage the
research and development of pharmaceutical products addressing conditions with significant unmet medical need. These designations may
confer benefits such as additional interaction with regulatory authorities, a potentially accelerated regulatory pathway and priority
review. There can be no assurance that we will successfully obtain such designation for any of our other product candidates. In addition,
while such designations could expedite the development or approval process, they generally do not change the standards for approval.
Even if we obtain such designations for one or more of our product candidates, there can be no assurance that we will realize their intended
benefits.
For
example, we may seek a Breakthrough Therapy Designation for one or more of our product candidates. A breakthrough therapy is defined
as a therapy that is intended, alone or in combination with one or more other therapies, to treat a serious or life-threatening disease
or condition, if preliminary clinical evidence indicates that the therapy may demonstrate substantial improvement over existing therapies
on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. For therapies
that have been designated as breakthrough therapies, interaction and communication between the FDA and the sponsor of the trial can help
to identify the most efficient path for clinical development while minimizing the number of patients placed in ineffective control regimens.
Therapies designated as breakthrough therapies by the FDA are also eligible for accelerated approval. Designation as a breakthrough therapy
is within the discretion of the FDA. Accordingly, even if we believe one of our product candidates meets the criteria for designation
as a breakthrough therapy, the FDA may disagree and instead determine not to make such designation. In any event, the receipt of a Breakthrough
Therapy Designation for a product candidate may not result in a faster development process, review or approval compared to therapies
considered for approval under conventional FDA procedures and does not assure ultimate approval by the FDA. In addition, even if one
or more of our product candidates qualify as breakthrough therapies, the FDA may later decide that such product candidates no longer
meet the conditions for qualification.
We
may also seek Fast Track Designation for some of our product candidates. If a therapy is intended for the treatment of a serious or life-threatening
condition and the therapy demonstrates the potential to address unmet medical needs for this condition, the therapy sponsor may apply
for Fast Track Designation. The FDA has broad discretion whether or not to grant this designation, so even if we believe a particular
product candidate is eligible for this designation, there can be no assurance that the FDA would decide to grant it. Even if we do receive
Fast Track Designation, we may not experience a faster development process, review or approval compared to conventional FDA procedures,
and receiving a Fast Track Designation does not provide assurance of ultimate FDA approval. The FDA may withdraw Fast Track Designation
if it believes that the designation is no longer supported by data from our clinical development program.
We
may seek priority review designation for one or more of our product candidates, but we might not receive such designation, and even if
we do, such designation may not lead to a faster regulatory review or approval process.
If
the FDA determines that a product candidate offers a treatment for a serious condition and, if approved, the product would provide a
significant improvement in safety or effectiveness, the FDA may designate the product candidate for priority review. A priority review
designation means that the goal for the FDA to review an application is six months, rather than the standard review period of ten months.
We may request priority review for our product candidates. The FDA has broad discretion with respect to whether or not to grant priority
review status to a product candidate, so even if we believe a particular product candidate is eligible for such designation or status,
in particular if such product candidate has received a Breakthrough Therapy Designation, the FDA may decide not to grant it. Moreover,
a priority review designation does not result in expedited development and does not necessarily result in expedited regulatory review
or approval process or necessarily confer any advantage with respect to approval compared to conventional FDA procedures. Receiving priority
review from the FDA does not guarantee approval within the six-month review cycle or at all.
Obtaining
and maintaining marketing approval of our current and future product candidates in one jurisdiction does not mean that we will be successful
in obtaining marketing approval of our current and future product candidates in other jurisdictions.
Obtaining
and maintaining marketing approval of our current and future product candidates in one jurisdiction does not guarantee that we will be
able to obtain or maintain marketing approval in any other jurisdiction, while a failure or delay in obtaining marketing approval in
one jurisdiction may have a negative effect on the marketing approval process in others. For example, even if the FDA grants marketing
approval of a product candidate, comparable regulatory authorities in foreign jurisdictions must also approve the manufacturing, marketing
and promotion of the product candidate in those countries. Approval procedures vary among jurisdictions and can involve requirements
and administrative review periods different from, and greater than, those in the United States, including additional preclinical studies
or clinical trials as clinical studies conducted in one jurisdiction may not be accepted by regulatory authorities in other jurisdictions.
In many jurisdictions outside the United States, a product candidate must be approved for reimbursement before it can be approved for
sale in that jurisdiction. In some cases, the price that we intend to charge for our products is also subject to approval. We do not
have experience in obtaining reimbursement or pricing approvals in international markets.
Obtaining
marketing approvals and compliance with regulatory requirements could result in significant delays, difficulties and costs for us and
could delay or prevent the introduction of our products in certain countries outside of the United States. If we fail to comply with
the regulatory requirements in international markets and/or receive applicable marketing approvals, our target market will be reduced
and our ability to realize the full market potential of our product candidates will be harmed.
Risks
Related to Commercialization of Our Product Candidates and Other Regulatory Compliance Matters
Even
if we complete the necessary preclinical studies and clinical trials, the marketing approval process is expensive, time consuming and
uncertain and may prevent us or any collaborators from obtaining approvals for the commercialization of some or all of our product candidates.
As a result, we cannot predict when or if, and in which territories, we, or any collaborators, will obtain marketing approval to commercialize
a product candidate.
The
process of obtaining marketing approvals, both in the United States and abroad, is lengthy, expensive and uncertain. It may take many
years, if approval is obtained at all, and can vary substantially based upon a variety of factors, including the type, complexity and
novelty of the product candidates involved. Securing marketing approval requires the submission of extensive preclinical and clinical
data and supporting information to regulatory authorities for each therapeutic indication to establish the product candidate’s
safety and efficacy. Securing marketing approval also requires the submission of information about the product manufacturing process
to, and inspection of manufacturing facilities by, the regulatory authorities. The FDA or other regulatory authorities may determine
that our product candidates are not safe and effective, only moderately effective or have undesirable or unintended side effects, toxicities
or other characteristics that preclude our obtaining marketing approval or prevent or limit commercial use. Any marketing approval we
ultimately obtain may be limited or subject to restrictions or post-approval commitments that render the approved product not commercially
viable.
In
addition, changes in marketing approval policies during the development period, changes in or the enactment or promulgation of additional
statutes, regulations or guidance or changes in regulatory review for each submitted product application, may cause delays in the approval
or rejection of an application. Regulatory authorities have substantial discretion in the approval process and may refuse to accept any
application or may decide that our data are insufficient for approval and require additional preclinical, clinical or other studies.
Varying interpretations of the data obtained from preclinical and clinical testing could delay, limit or prevent marketing approval of
a product candidate. We cannot commercialize a product until the appropriate regulatory authorities have reviewed and approved the product
candidate. Even if our product candidates demonstrate safety and efficacy in clinical trials, the regulatory agencies may not complete
their review processes in a timely manner, or we may not be able to obtain regulatory approval. Additional delays may result if an FDA
Advisory Committee or other regulatory authority recommends non-approval or restrictions on approval. In addition, we may experience
delays or rejections based upon additional government regulation from future legislation or administrative action, or changes in regulatory
agency policy during the period of product development, clinical trials and the review process. Any marketing approval we ultimately
obtain may be limited or subject to restrictions or post-approval commitments that render the approved product commercially unviable.
Moreover,
principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time and receive compensation
in connection with such services. Under certain circumstances, we may be required to report some of these relationships to the FDA or
other regulatory authority. The FDA or other regulatory authority may conclude that a financial relationship between us and a principal
investigator has created a conflict of interest or otherwise affected interpretation of the study. The FDA or other regulatory authority
may therefore question the integrity of the data generated at the applicable clinical trial site and the utility of the clinical trial
itself may be jeopardized. This could result in a delay in approval, or rejection, of our marketing applications by the FDA or other
regulatory authority, as the case may be, and may ultimately lead to the denial of marketing approval of one or more of our product candidates.
In
addition, regulatory agencies may not approve the labeling claims that are necessary or desirable for the successful commercialization
of our product candidates. For example, regulatory agencies may approve a product candidate for fewer or more limited indications than
requested or may grant approval subject to the performance of post-marketing studies. Regulators may approve a product candidate for
a smaller patient population, a different drug formulation or a different manufacturing process, than we are seeking. If we are unable
to obtain necessary regulatory approvals, or more limited regulatory approvals than we expect, our business, prospects, financial condition
and results of operations may suffer.
Any
delay in obtaining or failure to obtain required approvals could negatively impact our ability to generate revenue from the particular
product candidate, which likely would result in significant harm to our financial position and adversely impact the price of our common
stock.
We
currently have no marketing, sales or distribution infrastructure with respect to our product candidates. If we are unable to develop
our sales, marketing and distribution capability on our own or through collaborations with marketing partners, we will not be successful
in commercializing our product candidates.
We
currently have no marketing, sales or distribution capabilities and have limited sales or marketing experience within our organization.
If one or more of our product candidates is approved, we intend either to establish a sales and marketing organization with technical
expertise and supporting distribution capabilities to commercialize that product candidate, or to outsource this function to a third
party. There are risks involved with either establishing our own sales and marketing capabilities and entering into arrangements with
third parties to perform these services.
Recruiting
and training an internal commercial organization is expensive and time consuming and could delay any product launch. Some or all of these
costs may be incurred in advance of any approval of any of our product candidates. If the commercial launch of a product candidate for
which we recruit a sales force and establish marketing capabilities is delayed or does not occur for any reason, we would have prematurely
or unnecessarily incurred these commercialization expenses. This may be costly and our investment would be lost if we cannot retain or
reposition our sales and marketing personnel. In addition, we may not be able to hire a sales force in the United States or other target
market that is sufficient in size or has adequate expertise in the medical markets that we intend to target.
Factors
that may inhibit our efforts to commercialize our product candidates on our own include:
●
the inability to recruit, train and retain adequate numbers of effective sales and marketing personnel;
●
the inability of sales personnel to obtain access to physicians or persuade adequate numbers of physicians to prescribe any future product
that we may develop;
●
the lack of complementary treatments to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies
with more extensive product lines; and
●
unforeseen costs and expenses associated with creating an independent sales and marketing organization.
If
we enter into arrangements with third parties to perform sales, marketing and distribution services, our product revenue or the profitability
to us from these revenue streams is likely to be lower than if we were to market and sell any product candidates that we develop ourselves.
In addition, we may not be successful in entering into arrangements with third parties to sell and market our product candidates or may
be unable to do so on terms that are favorable to us. We likely will have little control over such third parties and any of them may
fail to devote the necessary resources and attention to sell and market our product candidates effectively. If we do not establish sales
and marketing capabilities successfully, either on our own or in collaboration with third parties, we may not be successful in commercializing
our product candidates.
The
market opportunities for any current or future product candidate we develop, if and when approved, may be limited to those patients who
are ineligible for established therapies or for whom prior therapies have failed, and therefore may be small.
Cancer
therapies are sometimes characterized as first-line, second-line, or third-line, and the FDA often approves new therapies initially only
for third-line use. When cancer is detected early enough, first-line therapy, usually chemotherapy, hormone therapy, surgery, radiation
therapy, immunotherapy or a combination of these, is sometimes adequate to cure the cancer or prolong life without a cure. Second- and
third-line therapies are administered to patients when prior therapy is not effective. We may initially seek approval of SON-080 and
any other product candidates we develop as a therapy for patients who have received one or more prior treatments. Subsequently, for those
products that prove to be sufficiently beneficial, if any, we would expect to seek approval potentially as a first-line therapy, but
there is no guarantee that product candidates we develop, even if approved, would be approved for first-line therapy, and, prior to any
such approvals, we may have to conduct additional clinical trials.
The
number of patients who have the cancers we are targeting may turn out to be lower than expected. Additionally, the potentially addressable
patient population for our current programs or future product candidates may be limited, if and when approved. Even if we obtain significant
market share for any product candidate, if and when approved, if the potential target populations are small, we may never achieve profitability
without obtaining marketing approval for additional indications, including use as first- or second-line therapy.
Even
if we receive marketing approval of a product candidate, we will be subject to ongoing regulatory obligations and continued regulatory
review, which may result in significant additional expense and we may be subject to penalties if we fail to comply with regulatory requirements
or experience unanticipated problems with our products, if approved.
Any
marketing approvals that we receive for any current or future product candidate may be subject to limitations on the approved indicated
uses for which the product may be marketed or the conditions of approval, or contain requirements for potentially costly post-market
testing and surveillance to monitor the safety and efficacy of the product candidate. The FDA may also require a Risk Evaluation and
Mitigation Strategy, or REMS, as a condition of approval of any product candidate, which could include requirements for a medication
guide, physician communication plans or additional elements to ensure safe use, such as restricted distribution methods, patient registries
and other risk minimization tools. If the FDA or a comparable foreign regulatory authority approves a product candidate, the manufacturing
processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion, import and export and record
keeping for the product candidate will be subject to extensive and ongoing regulatory requirements. These requirements include, among
others, prohibitions on the promotion of an approved product for uses not included in the product’s approved labeling, submissions
of safety and other post-marketing information and reports, registration, as well as continued compliance with current Good Manufacturing
Practice, or cGMP, and Good Clinical Practice, or GCP, for any clinical trials that we conduct post-approval. Later discovery of previously
unknown problems with any approved candidate, including adverse events of unanticipated severity or frequency, or with our third-party
manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
●
restrictions on the labeling, distribution, marketing or manufacturing of the product, withdrawal of the product from the market, or
product recalls;
●
untitled and warning letters, or holds on clinical trials;
●
refusal by the FDA to approve pending applications or supplements to approved applications we filed or suspension or revocation of license
approvals;
●
requirements to conduct post-marketing studies or clinical trials;
●
restrictions on coverage by third-party payors;
●
fines, restitution or disgorgement of profits or revenues;
●
suspension or withdrawal of marketing approvals;
●
product seizure or detention, or refusal to permit the import or export of the product; and
●
injunctions or the imposition of civil or criminal penalties.
The
FDA’s and other regulatory authorities’ policies may change and additional government regulations may be enacted that could
prevent, limit or delay marketing approval of a product. We cannot predict the likelihood, nature or extent of government regulation
that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt
to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance,
we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability.
We
face significant competition and if our competitors develop and market products that are more effective, safer or less expensive than
the product candidates we develop, our commercial opportunities will be negatively impacted.
The
life sciences industry is highly competitive. We are currently developing therapeutics that will compete, if approved, with other products
and therapies that currently exist, are being developed or will in the future be developed, some of which we may not currently be aware.
We
have competitors both in the United States and internationally, including major multinational pharmaceutical companies, established biotechnology
companies, specialty pharmaceutical companies, universities and other research institutions. Many of our competitors have significantly
greater financial, manufacturing, marketing, product development, technical and human resources than we do. Large pharmaceutical companies,
in particular, have extensive experience in clinical testing, obtaining marketing approvals, recruiting patients and manufacturing pharmaceutical
products. These companies also have significantly greater research and marketing capabilities than we do and may also have products that
have been approved or are in late stages of development, and collaborative arrangements in our target markets with leading companies
and research institutions. Established pharmaceutical companies may also invest heavily to accelerate discovery and development of novel
compounds or to in-license novel compounds that could make the product candidates that we develop obsolete. Mergers and acquisitions
in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our
competitors. As a result of all of these factors, our competitors may succeed in obtaining patent protection and/or marketing approval
or discovering, developing and commercializing products in our field before we do.
There
is a large number of companies developing or marketing treatments for cancer, including many major pharmaceutical and biotechnology companies.
These treatments consist both of small molecule drug products, such as traditional chemotherapy, as well as novel immunotherapies. For
example, a number of multinational companies as well as large biotechnology companies, including Astellas Pharma Inc., Seattle Genetics,
Inc., AstraZeneca, and GlaxoSmithKline plc, are developing programs for the targets that we are exploring for our pipeline programs.
Our
commercial opportunities could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective,
have fewer or less severe effects, are more convenient, have a broader label, are marketed more effectively, are reimbursed or are less
expensive than any products that we may develop. Our competitors also may obtain FDA, EMA or other marketing approval for their products
more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before
we are able to enter the market. Even if the product candidate we develop achieve marketing approval, they may be priced at a significant
premium over competitive products if any have been approved by then, resulting in reduced competitiveness.
Smaller
and other early stage companies may also prove to be significant competitors. These third parties compete with us in recruiting and retaining
qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well
as in acquiring technologies complementary to, or necessary for, our programs. In addition, the biopharmaceutical industry is characterized
by rapid technological change. If we fail to stay at the forefront of technological change, we may be unable to compete effectively.
Technological advances or products developed by our competitors may render our product candidates obsolete, less competitive or not economical.
The
commercial success of any current or future product candidate will depend upon the degree of market acceptance by physicians, patients,
payors and others in the medical community.
We
have never commercialized a product, and even if we obtain any regulatory approval for our product candidates, the commercial success
of our product candidates will depend in part on the medical community, patients, and payors accepting our product candidates as effective,
safe and cost-effective. Any product that we bring to the market may not gain market acceptance by physicians, patients, payors and others
in the medical community. Physicians are often reluctant to switch their patients from existing therapies even when new and potentially
more effective or convenient treatments enter the market. Further, patients often acclimate to the therapy that they are currently taking
and do not want to switch unless their physicians recommend switching products or they are required to switch therapies due to lack of
reimbursement for existing therapies.
The
degree of market acceptance of these product candidates, if approved for commercial sale, will depend on a number of factors, including:
| ● |
the
potential efficacy and potential advantages over alternative treatments; |
| |
|
| ● |
the
frequency and severity of any side effects, including any limitations or warnings contained in a product’s approved labeling; |
| |
|
| ● |
the
frequency and severity of any side effects resulting from follow-up requirements for the administration of our product candidates; |
| |
|
| ● |
the
relative convenience and ease of administration; |
| |
|
| ● |
the
willingness of the target patient population to try new therapies and of physicians to prescribe these therapies; |
| |
|
| ● |
the
strength of marketing and distribution support and timing of market introduction of competitive products; |
| |
|
| ● |
publicity
concerning our products or competing products and treatments; and |
| |
|
| ● |
sufficient
third-party insurance coverage and adequate reimbursement. |
Even
if a product candidate displays a favorable efficacy and safety profile in preclinical studies and clinical trials, market acceptance
of the product, if approved for commercial sale, will not be known until after it is launched. Our efforts to educate the medical community
and payors on the benefits of our product candidates may require significant resources and may never be successful. Such efforts to educate
the marketplace may require more resources than are required by the conventional technologies marketed by our competitors, particularly
due to the novelty of our Sonnet approach. If these products do not achieve an adequate level of acceptance, we may not generate
significant product revenue and may not become profitable.
If
the market opportunities for our product candidates are smaller than we believe they are, our product revenues may be adversely affected
and our business may suffer.
We
currently focus our research and product development on treatments for oncology indications and our product FHAB candidates
are designed to target solid tumors. Our understanding of both the number of people who have these diseases, as well as the subset of
people with these diseases who have the potential to benefit from treatment with our product candidates, are based on estimates. These
estimates may prove to be incorrect and new studies may reduce the estimated incidence or prevalence of these diseases. Patient identification
efforts also influence the ability to address a patient population. If efforts in patient identification are unsuccessful or less impactful
than anticipated, we may not address the entirety of the opportunity we are seeking.
The
insurance coverage and reimbursement status of newly-approved products is uncertain. Failure to obtain or maintain adequate coverage
and reimbursement for any of our product candidates, if approved, could limit our ability to market those products and decrease our ability
to generate revenue.
We
expect the cost of our product candidates to be substantial, when and if they achieve market approval. The availability and extent of
reimbursement by governmental and private payors is essential for most patients to be able to afford expensive treatments. Sales of our
product candidates will depend substantially, both domestically and abroad, on the extent to which the costs of our product candidates
will be paid by private payors, such as private health coverage insurers, health maintenance, managed care, pharmacy benefit and similar
healthcare management organizations, or reimbursed by government health care programs, such as Medicare and Medicaid. We may not be able
to provide data sufficient to gain acceptance with respect to coverage and reimbursement. If reimbursement is not available, or is available
only at limited levels, we may not be able to successfully commercialize our product candidates, even if approved. Even if coverage is
provided, the approved reimbursement amount may not be high enough to allow us to establish or maintain pricing sufficient to realize
a sufficient return on our investment.
There
is significant uncertainty related to the insurance coverage and reimbursement of newly approved products. In the United States, the
principal decisions about coverage and reimbursement for new medicines are typically made by the Centers for Medicare & Medicaid
Services, or CMS, an agency within the U.S. Department of Health and Human Services, as the CMS decides whether and to what extent a
new medicine will be covered and reimbursed under Medicare. Private payors tend to follow CMS to a substantial degree. It is difficult
to predict what CMS will decide with respect to coverage and reimbursement for novel products such as ours, as there is no body of established
practices and precedents for these new products. Coverage and reimbursement by a third-party payor may depend upon a number of factors,
including the third-party payor’s determination that use of a product is: (1) a covered benefit under its health plan; (2) safe,
effective and medically necessary; (3) appropriate for the specific patient; (4) cost-effective; and (5) neither experimental nor investigational.
In the United States, no uniform policy of coverage and reimbursement for products exists among third-party payors. As a result, the
coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support
for the use of our products to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently
or obtained in the first instance. Even if we obtain coverage for a given product, the resulting reimbursement payment rates might not
be adequate for us to achieve or sustain profitability or may require co-payments that patients find unacceptably high. Third-party payors
may limit coverage to specific drug products on an approved list, also known as a formulary, which might not include all of the approved
drugs for a particular indication.
Additionally,
third-party payors may not cover, or provide adequate reimbursement for, long-term follow-up evaluations required following the use of
product candidates. Patients are unlikely to use our product candidates unless coverage is provided and reimbursement is adequate to
cover a significant portion of the cost of our product candidates. Because our product candidates may have a higher cost of goods than
conventional therapies, and may require long-term follow-up evaluations, the risk that coverage and reimbursement rates may be inadequate
for us to achieve profitability may be greater. There is significant uncertainty related to insurance coverage and reimbursement of newly
approved products. It is difficult to predict at this time what third-party payors will decide with respect to the coverage and reimbursement
for our product candidates.
Moreover,
increasing efforts by governmental and third-party payors in the United States and abroad to cap or reduce healthcare costs may cause
such organizations to limit both coverage and the level of reimbursement for newly approved products and, as a result, they may not cover
or provide adequate payment for our product candidates. There has been increasing legislative and enforcement interest in the United
States with respect to specialty drug pricing practices. Specifically, there have been several recent U.S. Congressional inquiries and
proposed federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription
drugs under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement
methodologies for drugs. We expect to experience pricing pressures in connection with the sale of any of our product candidates due to
the trend toward managed healthcare, the increasing influence of health maintenance organizations, cost containment initiatives and additional
legislative changes.
Outside
the United States, certain countries, including a number of member states of the European Union, set prices and reimbursement for pharmaceutical
products, or medicinal products, as they are commonly referred to in the European Union. These countries have broad discretion in setting
prices and we cannot be sure that such prices and reimbursement will be acceptable to us or our collaborators. If the regulatory authorities
in these jurisdictions set prices or reimbursement levels that are not commercially attractive for us or our collaborators, our revenues
from sales by us or our collaborators, and the potential profitability of our drug products, in those countries would be negatively affected.
An increasing number of countries are taking initiatives to attempt to reduce large budget deficits by focusing cost-cutting efforts
on pharmaceuticals for their state-run health care systems. These international price control efforts have impacted all regions of the
world, but have been most drastic in the European Union. Additionally, some countries require approval of the sale price of a product
before it can be lawfully marketed. In many countries, the pricing review period begins after marketing or product licensing approval
is granted. To obtain reimbursement or pricing approval in some countries, we, or any collaborators, may be required to conduct a clinical
trial that compares the cost-effectiveness of our product to other available therapies. As a result, we might obtain marketing approval
for a product in a particular country, but then may experience delays in the reimbursement approval of our product or be subject to price
regulations that would delay our commercial launch of the product, possibly for lengthy time periods, which could negatively impact the
revenues we are able to generate from the sale of the product in that particular country.
Moreover,
efforts by governments and payors, in the United States and abroad, to cap or reduce healthcare costs may cause such organizations to
limit both coverage and level of reimbursement for new products approved and, as a result, they may not cover or provide adequate reimbursement
for our product candidates. There has been increasing legislative and enforcement interest in the United States with respect to specialty
drug practices. Specifically, there have been several recent U.S. Congressional inquiries and proposed federal and state legislation
designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, review
the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs.
We expect to experience pricing pressures in connection with the sale of any of our product candidates, due to the trend toward managed
healthcare, the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on
healthcare costs in general, particularly prescription drugs and other treatments, has become very intense. As a result, increasingly
high barriers are being erected to the entry of new products. If reimbursement of our products is unavailable or limited in scope or
amount, or if pricing is set at unsatisfactory levels, our business could be harmed.
If
the FDA or comparable foreign regulatory authorities approve generic versions of any of our product candidates that receive marketing
approval, or such authorities do not grant such products appropriate periods of data exclusivity before approving generic versions of
such products, the sales of such products could be adversely affected.
In
the United States, manufacturers may seek approval of biosimilar versions of biologics approved by the FDA under a BLA through submission
of abbreviated biologic license applications, or ABLAs. In support of an ABLA, a biosimilar manufacturer generally must show that its
product is similar to the original biologic product. Biosimilar products may be less costly to bring to market than the original biologic
and companies that produce biosimilar products are sometimes able to offer them at lower prices. Thus, following the introduction of
a biosimilar product, a significant percentage of the sales of the original biologic may be lost to the biosimilar product, and the price
of the original biologic product may be lowered.
The
FDA may not accept for review or approve an ABLA for a biosimilar product until any applicable period of non-patent exclusivity for the
original biologic has expired. The Public Health Service (PHS) Act provides a period of twelve years of non-patent exclusivity for a
biologic approved under a BLA.
Competition
that our products may face from biosimilar versions of our products could negatively impact our future revenue, profitability and cash
flows and substantially limit our ability to obtain a return on our investments in those product candidates.
We
may be subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, false claims laws health information privacy
and security laws, and other health care laws and regulations. If we are unable to comply, or have not fully complied, with such laws,
we could face substantial penalties.
If
we obtain FDA approval for any of our product candidates and begin commercializing those products in the United States, our operations
will be directly, or indirectly through our prescribers, customers and purchasers, subject to various federal and state fraud and abuse
laws and regulations, including, without limitation, the federal Health Care Program Anti-Kickback Statute, or Anti-Kickback Statute,
the federal civil and criminal False Claims Act and Physician Payments Sunshine Act and regulations. These laws will impact, among other
things, our proposed sales, marketing and educational programs. In addition, we may be subject to patient privacy laws by both the federal
government and the states in which we conduct our business. The laws that will affect our operations include, but are not limited to:
| ● |
the
Anti-Kickback Statute, which prohibits, among other things, persons or entities from knowingly and willfully soliciting, receiving,
offering or paying any remuneration (including any kickback, bribe or rebate), directly or indirectly, overtly or covertly, in cash
or in kind, to induce, or in return for, either the referral of an individual, or the purchase, lease, order, arrangement, or recommendation
of any good, facility, item or service for which payment may be made, in whole or in part, under a federal healthcare program, such
as the Medicare and Medicaid programs. “Remuneration” has been interpreted broadly to include anything of value. A person
or entity does not need to have actual knowledge of the Anti-Kickback Statute or specific intent to violate it to have committed
a violation. In addition, the government may assert that a claim including items or services resulting from a violation of the Anti-Kickback
Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act, or FCA, or federal civil money penalties.
The Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and
prescribers, purchasers, and formulary managers on the other. There are a number of statutory exceptions and regulatory safe harbors
protecting some common activities from prosecution; |
| ● |
the
federal civil and criminal false claims laws and civil monetary penalty laws, including the FCA, which impose criminal and civil
penalties against individuals or entities for, among other things: knowingly presenting, or causing to be presented, to the federal
government, claims for payment that are false or fraudulent; knowingly making, using or causing to be made or used, a false statement
of record material to a false or fraudulent claim or obligation to pay or transmit money or property to the federal government. Manufacturers
can be held liable under the FCA even when they do not submit claims directly to government payors if they are deemed to “cause”
the submission of false or fraudulent claims. The FCA also permits a private individual acting as a “whistleblower” to
bring actions on behalf of the federal government alleging violations of the FCA and to share in any monetary recovery; |
| |
|
| ● |
the
beneficiary inducement provisions of the CMP Law, which prohibits, among other things, the offering or giving of remuneration, which
includes, without limitation, any transfer of items or services for free or for less than fair market value (with limited exceptions),
to a Medicare or Medicaid beneficiary that the person knows or should know is likely to influence the beneficiary’s selection
of a particular supplier of items or services reimbursable by a federal or state governmental program; |
| |
|
| ● |
the
federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created new federal criminal statutes that prohibit
a person from knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or
obtain, by means of false or fraudulent pretenses, representations or promises, any of the money or property owned by, or under the
custody or control of, any healthcare benefit program, regardless of the payor (e.g., public or private) and knowingly and willfully
falsifying, concealing or covering up by any trick or device a material fact or making any materially false, fictitious, or fraudulent
statements or representations in connection with the delivery of, or payment for, healthcare benefits, items or services relating
to healthcare matters; similar to the Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute
or specific intent to violate it in order to have committed a violation; |
| |
|
| ● |
HIPAA,
as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, and their respective implementing regulations,
which impose requirements on certain healthcare providers, health plans, and healthcare clearinghouses, known as covered entities,
as well as their respective business associates, individuals and entities that perform services on their behalf that involve the
use or disclosure of individually identifiable health information, relating to the privacy, security and transmission of individually
identifiable health information; |
| |
|
| ● |
the
U.S. federal transparency requirements under the Patient Protection and Affordable Care Act, as amended by the Health Care and Education
Reconciliation Act, or collectively, ACA, including the provision commonly referred to as the Physician Payments Sunshine Act, which
requires applicable manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare,
Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to CMS information related
to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors)
and teaching hospitals, as well as ownership and investment interests held by the physicians described above and their immediate
family members. Beginning in 2022, applicable manufacturers also will be required to report information regarding payments and transfers
of value provided to physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists, and certified
nurse-midwives; |
| |
|
| ● |
federal
government price reporting laws, which require us to calculate and report complex pricing metrics in an accurate and timely manner
to government programs; and |
| |
|
| ● |
federal
consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm
consumers. |
Additionally,
we are subject to state and foreign equivalents of each of the healthcare laws and regulations described above, among others, some of
which may be broader in scope and may apply regardless of the payer. Many U.S. states have adopted laws similar to the Anti-Kickback
Statute and False Claims Act, and may apply to our business practices, including, but not limited to, research, distribution, sales or
marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental payors, including private insurers.
In addition, some states have passed laws that require pharmaceutical companies to comply with the April 2003 Office of Inspector General
Compliance Program Guidance for Pharmaceutical Manufacturers and/or the Pharmaceutical Research and Manufacturers of America’s
Code on Interactions with Healthcare Professionals. Several states also impose other marketing restrictions or require pharmaceutical
companies to make marketing or price disclosures to the state. There are ambiguities as to what is required to comply with these state
requirements and if we fail to comply with an applicable state law requirement we could be subject to penalties. Finally, there are state
and foreign laws governing the privacy and security of health information, many of which differ from each other in significant ways and
often are not preempted by HIPAA, thus complicating compliance efforts.
Because
of the breadth of these laws and the narrowness of the statutory exceptions and regulatory safe harbors available, it is possible that
some of our business activities could be subject to challenge under one or more of such laws. Law enforcement authorities are increasingly
focused on enforcing fraud and abuse laws, and it is possible that some of our practices may be challenged under these laws. Efforts
to ensure that our current and future business arrangements with third parties, and our business generally, will comply with applicable
healthcare laws and regulations will involve substantial costs. If our operations, including our arrangements with physicians and other
healthcare providers, some of whom receive stock options as compensation for services provided, are found to be in violation of any of
such laws or any other governmental regulations that apply to us, we may be subject to penalties, including, without limitation, administrative,
civil and criminal penalties, damages, fines, disgorgement, contractual damages, reputational harm, diminished profits and future earnings,
the curtailment or restructuring of our operations, exclusion from participation in federal and state healthcare programs (such as Medicare
and Medicaid), additional reporting requirements and/or oversight if we become subject to a corporate integrity agreement or similar
agreement to resolve allegations of non-compliance with these laws, and individual imprisonment, any of which could adversely affect
our ability to operate our business and our financial results. Any action for violation of these laws, even if successfully defended,
could cause a pharmaceutical manufacturer to incur significant legal expenses and divert management’s attention from the operation
of the business. Prohibitions or restrictions on sales or withdrawal of future marketed products could materially affect business in
an adverse way.
Healthcare
legislative reform measures and constraints on national budget social security systems may have a material adverse effect on our business
and results of operations.
Payors,
whether domestic or foreign, or governmental or private, are developing increasingly sophisticated methods of controlling healthcare
costs and those methods are not always specifically adapted for new technologies such as those we are developing. In both the United
States and certain foreign jurisdictions, there have been a number of legislative and regulatory changes to the health care system that
could impact our ability to sell our products profitably. In particular, in the United States, the ACA was enacted in 2010 which, among
other things, subjects biologic products to potential competition by lower-cost biosimilars; addresses a new methodology by which rebates
owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted
or injected; increases the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program; extends the Medicaid
Drug Rebate program to utilization of prescriptions of individuals enrolled in Medicaid managed care organizations; subjects manufacturers
to new annual fees and taxes for certain branded prescription drugs; and provides incentives to programs that increase the federal government’s
comparative effectiveness research.
Since
its enactment, there have been judicial and Congressional challenges to certain aspects of the ACA, as well as recent efforts by the
current administration to repeal or replace certain aspects of the ACA. Further, since January 2017, President Trump has signed two Executive
Orders and other directives designed to delay the implementation of certain provision of the ACA or otherwise circumvent some of the
requirements for health insurance mandated by the ACA. In addition, CMS recently issued a final rule that will give states greater flexibility,
starting in 2020, in setting benchmarks for insurers in the individual and small group marketplaces, which may have the effect of relaxing
the essential health benefits required under the ACA for plans sold through such marketplaces.
Concurrently,
Congress has considered legislation that would repeal and replace all or part of the ACA. While Congress has not passed comprehensive
repeal legislation, two bills affecting the implementation of certain taxes under the ACA have been signed into law. The Tax Cuts and
Jobs Act of 2017, or TCJA, includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed
by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred
to as the “individual mandate.” Additionally, on January 22, 2018, President Trump signed a continuing resolution on appropriations
for fiscal year 2018 that delayed the implementation of certain ACA-mandated fees, including the so-called “Cadillac” tax
on certain high cost employer-sponsored insurance plans, the annual fee imposed on certain high cost employer-sponsored insurance plans,
the annual fee imposed on certain health insurance providers based on market share, and the medical device exercise tax on non-exempt
medical devices. Further, the Bipartisan Budget Act of 2018, or BBA, among other things, amends the ACA, effective January 1, 2019, to
increase from 50 percent to 70 percent the point-of-sale discount that is owed by pharmaceutical manufacturers who participate in Medicare
Part D and to close the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole.” More recently,
in July 2018, CMS published a final rule permitting further collections and payments to and from certain ACA qualified health plans and
health insurance issuers under the ACA risk adjustment program in response to the outcome of federal district court litigation regarding
the method CMS uses to determine this risk adjustment. Congress also could consider additional legislation to repeal or replace other
elements of the ACA. Thus, the full impact of the ACA, any law repealing or replacing elements of it, and the political uncertainty surrounding
any repeal or replacement legislation on our business remains unclear.
In
addition, other legislative changes have been proposed and adopted in the United States since the ACA was enacted. In August 2011, the
Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit
Reduction, tasked with recommending a targeted deficit reduction of at least $1.5 trillion for the years 2013 through 2021, was unable
to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes
aggregate reductions of Medicare payments to providers of 2% per fiscal year, which went into effect in April 2013, and due to subsequent
legislative amendments, including the BBA, will remain in effect through 2027 unless additional Congressional action is taken. In January
2013, the American Taxpayer Relief Act of 2012, was signed into law, which, among other things, further reduced Medicare payments to
several providers, including hospitals and cancer treatment centers, and increased the statute of limitations period for the government
to recover overpayments to providers from three to five years.
Also,
there has been heightened governmental scrutiny recently over the manner in which drug manufacturers set prices for their marketed products,
which has resulted in several Congressional inquiries and proposed and enacted federal and state legislation designed to, among other
things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform
government program reimbursement methodologies for drug products. For example, the current administration released a “Blueprint”
to lower drug prices and reduce out of pocket costs of drugs that contains additional proposals to increase manufacturer competition,
increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products
and reduce the out of pocket costs of drug products paid by consumers. For example, in November 2018, CMS issued a proposed rule for
comment that would, among other things, provide Medicare prescription drug plans under Part D more transparency in pricing and greater
flexibility to negotiate discounts for, and in certain circumstances exclude, drugs in the six “protected” formulary classes
and allow Medicare Advantage plans to use certain drug management tools such as step therapy for physician-administered drugs. Although
a number of these, and other proposed measures will require authorization through additional legislation to become effective, Congress
and the Trump administration have each indicated that it will continue to seek new legislative and/or administrative measures to control
drug costs.
There
have been, and likely will continue to be, legislative and regulatory proposals at the foreign, federal and state levels directed at
broadening the availability of healthcare and containing or lowering the cost of healthcare. We cannot predict the initiatives that may
be adopted in the future. The continuing efforts of these governments and other payors to contain or reduce costs of healthcare and/or
impose price controls may adversely affect:
● the demand for our product candidates, if we obtain regulatory approval;
● our ability to set a price that we believe is fair for our products;
● our ability to generate revenue and achieve or maintain profitability;
● the level of taxes that we are required to pay; and
● the availability of capital.
Any
denial in coverage or reduction in reimbursement from Medicare or other government programs may result in a similar denial or reduction
in payments from private payors, which may adversely affect our future profitability.
We
are subject to the U.S. Foreign Corrupt Practices Act of 1977, as amended, or the FCPA, and other anti-corruption laws, as well as export
control laws, import and customs laws, trade and economic sanctions laws and other laws governing our operations.
Our
operations are subject to anti-corruption laws, including the FCPA, the U.S. domestic bribery statute contained in 18 §201, the
U.S. Travel Act, and other anti-corruption laws that apply in countries where we do business. The Bribery Act, the FCPA and these other
laws generally prohibit us and our employees and intermediaries from authorizing, promising, offering, or providing, directly or indirectly,
improper or prohibited payments, or anything else of value, to government officials or other persons to obtain or retain business or
gain some other business advantage. Under the Bribery Act, we may also be liable for failing to prevent a person associated with us from
committing a bribery offense. We and our commercial partners operate in a number of jurisdictions that pose a high risk of potential
Bribery Act or FCPA violations, and we participate in collaborations and relationships with third parties whose corrupt or illegal activities
could potentially subject us to liability under the Bribery Act, FCPA or local anti-corruption laws, even if we do not explicitly authorize
or have actual knowledge of such activities. In addition, we cannot predict the nature, scope or effect of future regulatory requirements
to which our international operations might be subject or the manner in which existing laws might be administered or interpreted.
We
are also subject to other laws and regulations governing our international operations, including regulations administered by the governments
of the United Kingdom and the United States, and authorities in the European Union, including applicable export control regulations,
economic sanctions and embargoes on certain countries and persons, anti-money laundering laws, import and customs requirements and currency
exchange regulations, collectively referred to as the Trade Control laws.
There
is no assurance that we will be completely effective in ensuring our compliance with all applicable anti-corruption laws, including the
Bribery Act, the FCPA or other legal requirements, including Trade Control laws. If we are not in compliance with the Bribery Act, the
FCPA and other anti-corruption laws or Trade Control laws, we may be subject to criminal and civil penalties, disgorgement and other
sanctions and remedial measures, and legal expenses, which could have an adverse impact on our business, financial condition, results
of operations and liquidity. Likewise, any investigation of any potential violations of the Bribery Act, the FCPA, other anti-corruption
laws or Trade Control laws by United Kingdom, United States or other authorities could also have an adverse impact on our reputation,
our business, results of operations and financial condition.
If
we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur
costs that could have a material adverse effect on the success of our business.
We
are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the
handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable
materials, including chemicals and biological materials. Our operations also produce hazardous waste products. We generally contract
with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these
materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting
damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines
and penalties. Furthermore, environmental laws and regulations are complex, change frequently and have tended to become more stringent.
We cannot predict the impact of such changes and cannot be certain of our future compliance. In addition, we may incur substantial costs
in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations
may impair our research, development or production efforts. Failure to comply with these laws and regulations also may result in substantial
fines, penalties or other sanctions.
Although
we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting
from the use of hazardous materials or other work-related injuries, this insurance may not provide adequate coverage against potential
liabilities. In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws
and regulations. These current or future laws and regulations may impair our research, development or production efforts. Failure to
comply with these laws and regulations also may result in substantial fines, penalties or other sanctions or liabilities, which could
materially adversely affect our business, financial condition, results of operations and prospects.
Risks
Related to Our International Operations
As
one of our subsidiaries, Relief, is based outside of the United States, we are subject to economic, political, regulatory and other risks
associated with international operations.
As
Relief is based in the Switzerland, our business is subject to risks associated with conducting business outside of the United States.
Many of our suppliers and clinical trial relationships are located outside the United States. Accordingly, our future results could be
harmed by a variety of factors, including:
| ● |
economic
weakness, including inflation, or political instability in particular non-U.S. economies and markets; |
| |
|
| ● |
differing
and changing regulatory requirements for product approvals; |
| |
|
| ● |
differing
jurisdictions could present different issues for securing, maintaining or obtaining freedom to operate in such jurisdictions; |
| |
|
| ● |
potentially
reduced protection for intellectual property rights; |
| |
|
| ● |
difficulties
in compliance with different, complex and changing laws, regulations and court systems of multiple jurisdictions and compliance with
a wide variety of foreign laws, treaties and regulations; |
| |
|
| ● |
changes
in non-U.S. regulations and customs, tariffs and trade barriers; |
| |
|
| ● |
changes
in non-U.S. currency exchange rates of the pound sterling, U.S. dollar, euro and currency controls; |
| |
|
| ● |
trade
protection measures, import or export licensing requirements or other restrictive actions by governments; |
| |
|
| ● |
differing
reimbursement regimes and price controls in certain non-U.S. markets; |
| |
|
| ● |
negative
consequences from changes in tax laws; |
| |
|
| ● |
compliance
with tax, employment, immigration and labor laws for employees living or traveling abroad, including, for example, the variable tax
treatment in different jurisdictions of options granted under our share option schemes or equity incentive plans; |
| |
|
| ● |
workforce
uncertainty in countries where labor unrest is more common than in the United States; |
| |
|
| ● |
litigation
or administrative actions resulting from claims against us by current or former employees or consultants individually or as part
of class actions, including claims of wrongful terminations, discrimination, misclassification or other violations of labor law or
other alleged conduct; |
| |
|
| ● |
difficulties
associated with staffing and managing international operations, including differing labor relations; |
| |
|
| ● |
production
shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and |
| |
|
| ● |
business
interruptions resulting from geo-political actions, including war and terrorism, or natural disasters including earthquakes, typhoons,
floods and fires. |
European
data collection is governed by restrictive regulations governing the use, processing, and cross-border transfer of personal information.
The
collection and use of personal health data in the European Union is was governed by the provisions of the Data Protection Directive,
and which, as of May 25, 2018, has been superseded by the GDPR. These directives impose several requirements relating to the consent
of the individuals to whom the personal data relates, the information provided to the individuals, notification of data processing obligations
to the competent national data protection authorities and the security and confidentiality of the personal data. The Data Protection
Directive and GDPR also impose strict rules on the transfer of personal data out of the European Union to the United States. Failure
to comply with the requirements of the Data Protection Directive, the GDPR, and the related national data protection laws of the European
Union Member States may result in fines and other administrative penalties. While the Data Protection Directive did not apply to organizations
based outside the EU, the GDPR has expanded its reach to include any business, regardless of its location, that provides goods or services
to residents in the EU. This expansion would incorporate any potential clinical trial activities in EU member states. The GDPR imposes
strict requirements on controllers and processors of personal data, including special protections for “sensitive information”
which includes health and genetic information of data subjects residing in the EU. GDPR grants individuals the opportunity to object
to the processing of their personal information, allows them to request deletion of personal information in certain circumstances, and
provides the individual with an express right to seek legal remedies in the event the individual believes his or her rights have been
violated. Further, the GDPR imposes strict rules on the transfer of personal data out of the European Union to the United States or other
regions that have not been deemed to offer “adequate” privacy protections. Failure to comply with the requirements of the
GDPR and the related national data protection laws of the European Union Member States, which may deviate slightly from the GDPR, may
result in fines of up to 4% of global revenues, or € 20,000,000, whichever is greater. As a result of the implementation of the
GDPR, we may be required to put in place additional mechanisms ensuring compliance with the new data protection rules.
Risks
Related to Our Dependence on Third Parties
For
certain product candidates, we may depend on development and commercialization collaborators to develop and conduct clinical trials with,
obtain regulatory approvals for, and if approved, market and sell product candidates. If such collaborators fail to perform as expected,
the potential for us to generate future revenue from such product candidates would be significantly reduced and our business would be
harmed.
For
certain products candidates, we depend, or will depend, on our development and commercial collaborators to develop, conduct clinical
trials of, and, if approved, commercialize product candidates.
Our
current collaborations and any future collaborations that we enter into are subject to numerous risks, including:
| ● |
collaborators
have significant discretion in determining the efforts and resources that they will apply to the collaborations; |
| |
|
| ● |
collaborators
may not perform their obligations as expected or fail to fulfill their responsibilities in a timely manner, or at all; |
| |
|
| ● |
collaborators
may not pursue development and commercialization of any product candidates that achieve regulatory approval or may elect not to continue
or renew development or commercialization programs based on preclinical studies or clinical trial results, changes in the collaborators’
strategic focus or available funding or external factors, such as an acquisition, that divert resources or create competing priorities; |
| |
|
| ● |
collaborators
may delay preclinical studies or clinical trials, provide insufficient funding for clinical trials, stop a preclinical study or clinical
trial or abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product candidate for
clinical testing; |
| ● |
we
may not have access to, or may be restricted from disclosing, certain information regarding product candidates being developed or
commercialized under a collaboration and, consequently, may have limited ability to inform our shareholders about the status of such
product candidates; |
| |
|
| ● |
collaborators
could independently develop, or develop with third parties, products that compete directly or indirectly with our product candidates
if the collaborators believe that competitive products are more likely to be successfully developed or can be commercialized under
terms that are more economically attractive than ours; |
| |
|
| ● |
The
collaborations may not result in product candidates to develop and/or preclinical studies or clinical trials conducted as part of
the collaborations may not be successful; |
| |
|
| ● |
product
candidates developed with collaborators may be viewed by our collaborators as competitive with their own product candidates or products,
which may cause collaborators to stop commercialization of our product candidates; |
| |
|
| ● |
a
collaborator with marketing and distribution rights to one or more of our product candidates that achieve regulatory approval may
not commit sufficient resources to the marketing and distribution of any such product candidate; and |
| |
|
| ● |
collaborators
may not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite
litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential litigation. |
In
addition, certain collaboration and commercialization agreements provide our collaborators with rights to terminate such agreements,
which rights may or may not be subject to conditions, and which rights, if exercised, would adversely affect our product development
efforts and could make it difficult for us to attract new collaborators. In that event, we would likely be required to limit the size
and scope of efforts for the development and commercialization of such product candidates or products; we would likely be required to
seek additional financing to fund further development or identify alternative strategic collaborations; our potential to generate future
revenue from royalties and milestone payments from such product candidates or products would be significantly reduced, delayed or eliminated;
and it could have an adverse effect on our business and future growth prospects. Our rights to recover tangible and intangible assets
and intellectual property rights needed to advance a product candidate or product after termination of a collaboration may be limited
by contract, and we may not be able to advance a program post- termination.
If
conflicts arise with our development and commercialization collaborators or licensors, they may act in their own self-interest, which
may be adverse to the interests of our company.
We
may in the future experience disagreements with our development and commercialization collaborators or licensors. Conflicts may arise
in our collaboration and license arrangements with third parties due to one or more of the following:
| ● |
disputes
with respect to milestone, royalty and other payments that are believed due under the applicable agreements; |
| |
|
| ● |
disagreements
with respect to the ownership of intellectual property rights or scope of licenses; |
| |
|
| ● |
disagreements
with respect to the scope of any reporting obligations; |
| |
|
| ● |
unwillingness
on the part of a collaborator to keep us informed regarding the progress of its development and commercialization activities, or
to permit public disclosure of these activities; and |
| |
|
| ● |
disputes
with respect to a collaborator’s or our development or commercialization efforts with respect to our products and product candidates. |
Conflicts
with our development and commercialization collaborators or licensors could materially adversely affect our business, financial condition
or results of operations and future growth prospects.
We
will rely on third parties, including independent clinical investigators and CROs, to conduct and sponsor some of the clinical trials
of our product candidates. Any failure by a third party to meet its obligations with respect to the clinical development of our product
candidates may delay or impair our ability to obtain regulatory approval for our product candidates.
We
will be relying upon and plan to continue to rely upon third parties, including independent clinical investigators, academic partners,
regulatory affairs consultants and third-party CROs, to conduct our preclinical studies and clinical trials, including in some instances
sponsoring such clinical trials, and to engage with regulatory authorities and monitor and manage data for our ongoing preclinical and
clinical programs. Given the breadth of clinical therapeutic areas for which we believe our product candidates may have utility, we intend
to continue to rely on external service providers rather than build internal regulatory expertise.
Any
of these third parties may terminate their engagements with us under certain circumstances. We may not be able to enter into alternative
arrangements or do so on commercially reasonable terms. In addition, there is a natural transition period when a new contract research
organization begins work. As a result, delays would likely occur, which could negatively impact our ability to meet our expected clinical
development timelines and harm our business, financial condition and prospects.
We
remain responsible for ensuring that each of our preclinical studies and clinical trials is conducted in accordance with the applicable
protocol and legal, regulatory and scientific standards, and our reliance on these third parties does not relieve us of our regulatory
responsibilities. We and our third-party contractors and CROs are required to comply with GCP requirements, which are regulations and
guidelines enforced by the FDA, the Competent Authorities of the Member States of the European Economic Area, or EEA, and comparable
foreign regulatory authorities for all of our products in clinical development. Regulatory authorities enforce these GCP requirements
through periodic inspections of trial sponsors, principal investigators and trial sites. If we fail to exercise adequate oversight over
any of our academic partners or CROs or if we or any of our academic partners or CROs do not successfully carry out their contractual
duties or obligations, fail to meet expected deadlines, or if the quality or accuracy of the clinical data they obtain is compromised
due to the failure to adhere to our clinical protocols or regulatory requirements, or for any other reasons, the clinical data generated
in our clinical trials may be deemed unreliable and the FDA, the EMA or comparable foreign regulatory authorities may require us to perform
additional clinical trials before approving our marketing applications. We cannot assure you that upon a regulatory inspection of us,
our academic partners or our CROs or other third parties performing services in connection with our clinical trials, such regulatory
authority will determine that any of our clinical trials complies with GCP regulations. In addition, our clinical trials must be conducted
with product produced under applicable CGMP regulations. Our failure to comply with these regulations may require us to repeat clinical
trials, which would delay the regulatory approval process.
Furthermore,
the third parties conducting clinical trials on our behalf are not our employees, and except for remedies available to us under our agreements
with such contractors, we cannot control whether or not they devote sufficient time, skill and resources to our ongoing development programs.
These contractors may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting
clinical trials or other drug development activities, which could impede their ability to devote appropriate time to our clinical programs.
If these third parties, including clinical investigators, do not successfully carry out their contractual duties, meet expected deadlines
or conduct our clinical trials in accordance with regulatory requirements or our stated protocols, we may not be able to obtain, or may
be delayed in obtaining, marketing approvals for our product candidates. If that occurs, we will not be able to, or may be delayed in
our efforts to, successfully commercialize our product candidates.
In
addition, with respect to investigator-sponsored trials that may be conducted, we would not control the design or conduct of these trials,
and it is possible that the FDA or EMA will not view these investigator-sponsored trials as providing adequate support for future clinical
trials or market approval, whether controlled by us or third parties, for any one or more reasons, including elements of the design or
execution of the trials or safety concerns or other trial results. We expect that such arrangements will provide us certain information
rights with respect to the investigator-sponsored trials, including access to and the ability to use and reference the data, including
for our own regulatory submissions, resulting from the investigator-sponsored trials. However, we would not have control over the timing
and reporting of the data from investigator-sponsored trials, nor would we own the data from the investigator- sponsored trials. If we
are unable to confirm or replicate the results from the investigator-sponsored trials or if negative results are obtained, we would likely
be further delayed or prevented from advancing further clinical development. Further, if investigators or institutions breach their obligations
with respect to the clinical development of our product candidates, or if the data proves to be inadequate compared to the firsthand
knowledge we might have gained had the investigator-sponsored trials been sponsored and conducted by us, then our ability to design and
conduct any future clinical trials ourselves may be adversely affected. Additionally, the FDA or EMA may disagree with the sufficiency
of our right of reference to the preclinical, manufacturing or clinical data generated by these investigator-sponsored trials, or our
interpretation of preclinical, manufacturing or clinical data from these investigator-sponsored trials. If so, the FDA or EMA may require
us to obtain and submit additional preclinical, manufacturing, or clinical data.
We
intend to rely on third parties to manufacture product candidates, which increases the risk that we will not have sufficient quantities
of such product candidates or products or such quantities at an acceptable cost, which could delay, prevent or impair our development
or commercialization efforts.
We
do not own or operate manufacturing facilities for the production of clinical or commercial supplies of the product candidates that we
are developing or evaluating in our development programs. We have limited personnel with experience in drug manufacturing and lack the
resources and the capabilities to manufacture any of our product candidates on a clinical or commercial scale. We rely on third parties
for supply of our product candidates, and our strategy is to outsource all manufacturing of our product candidates and products to third
parties.
In
order to conduct clinical trials of product candidates, we will need to have them manufactured in potentially large quantities. Our third-
party manufacturers may be unable to successfully increase the manufacturing capacity for any of our product candidates in a timely or
cost- effective manner, or at all. In addition, quality issues may arise during scale-up activities and at any other time. For example,
ongoing data on the stability of our product candidates may shorten the expiry of our product candidates and lead to clinical trial material
supply shortages, and potentially clinical trial delays. If these third-party manufacturers are unable to successfully scale up the manufacture
of our product candidates in sufficient quality and quantity, the development, testing and clinical trials of that product candidate
may be delayed or infeasible, and regulatory approval or commercial launch of that product candidate may be delayed or not obtained,
which could significantly harm our business.
Our
use of new third-party manufacturers increases the risk of delays in production or insufficient supplies of our product candidates as
we transfer our manufacturing technology to these manufacturers and as they gain experience manufacturing our product candidates. Even
after a third-party manufacturer has gained significant experience in manufacturing our product candidates or even if we believe we have
succeeded in optimizing the manufacturing process, there can be no assurance that such manufacturer will produce sufficient quantities
of our product candidates in a timely manner or continuously over time, or at all.
We
may be delayed if we need to change the manufacturing process used by a third party. Further, if we change an approved manufacturing
process, then we may be delayed if the FDA or a comparable foreign authority needs to review the new manufacturing process before it
may be used.
We
operate an outsourced model for the manufacture of our product candidates, and contract with good manufacturing practice, or GMP, licensed
pharmaceutical contract development and manufacturing organizations. While we have engaged several third-party vendors to provide clinical
and non-clinical supplies and fill-finish services, we do not currently have any agreements with third-party manufacturers for long-term
commercial supplies. In the future, we may be unable to enter into agreements with third-party manufacturers for commercial supplies
of any product candidate that we develop, or may be unable to do so on acceptable terms. Even if we are able to establish and maintain
arrangements with third-party manufacturers, reliance on third- party manufacturers entails risks, including:
| ● |
reliance
on third-parties for manufacturing process development, regulatory compliance and quality assurance; |
| |
|
| ● |
limitations
on supply availability resulting from capacity and scheduling constraints of third-parties; |
| |
|
| ● |
the
possible breach of manufacturing agreements by third-parties because of factors beyond our control; and |
| |
|
| ● |
the
possible termination or non-renewal of the manufacturing agreements by the third-party, at a time that is costly or inconvenient
to us. |
Third-party
manufacturers may not be able to comply with cGMP requirements or similar regulatory requirements outside the United States. Our failure,
or the failure of our third-party manufacturers, to comply with applicable requirements could result in sanctions being imposed on us,
including fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls
of product candidates or products, operating restrictions and/or criminal prosecutions, any of which could significantly and adversely
affect supplies of our product candidates. In addition, some of the product candidates we intend to develop, including SON-080, use toxins
or other substances that can be produced only in specialized facilities with specific authorizations and permits, and there can be no
guarantee that we or our manufacturers can maintain such authorizations and permits. These specialized requirements may also limit the
number of potential manufacturers that we can engage to produce our product candidates, and impair any efforts to transition to replacement
manufacturers.
Our
future product candidates and any products that we may develop may compete with other product candidates and products for access to manufacturing
facilities. There are a limited number of manufacturers that operate under cGMP requirements that might be capable of manufacturing for
us.
If
the third parties that we engage to supply any materials or manufacture product for our preclinical tests and clinical trials should
cease to continue to do so for any reason, we likely would experience delays in advancing these tests and trials while we identify and
qualify replacement suppliers or manufacturers and we may be unable to obtain replacement supplies on terms that are favorable to us.
In addition, if we are not able to obtain adequate supplies of our product candidates or the substances used to manufacture them, it
will be more difficult for us to develop our product candidates and compete effectively.
Our
current and anticipated future dependence upon others for the manufacture of our product candidates may adversely affect our future profit
margins and our ability to develop product candidates and commercialize any products that receive marketing approval on a timely and
competitive basis.
Our
reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor will discover them
or that our trade secrets will be misappropriated or disclosed.
Because
we rely on third parties to manufacture our product candidates, and because we collaborate with various organizations and academic institutions
on the development of our product candidates, we must, at times, share trade secrets with them. We seek to protect our proprietary technology
in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research agreements,
consulting agreements or other similar agreements with our collaborators, advisors, employees and consultants prior to beginning research
or disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose our confidential
information, such as trade secrets.
Despite
the contractual provisions employed when working with third parties, the need to share trade secrets and other confidential information
increases the risk that such trade secrets become known by our competitors, are inadvertently incorporated into the technology of others,
or are disclosed or used in violation of these agreements. Given that our proprietary position is based, in part, on our know-how and
trade secrets, a competitor’s discovery of our trade secrets or other unauthorized use or disclosure would impair our competitive
position and may have a material adverse effect on our business.
In
addition, these agreements typically restrict the ability of our collaborators, advisors, employees and consultants to publish data potentially
relating to our trade secrets. Our academic collaborators typically have rights to publish data, provided that we are notified in advance
and may delay publication for a specified time in order to secure our intellectual property rights arising from the collaboration. In
other cases, publication rights are controlled exclusively by us, although in some cases we may share these rights with other parties.
Despite our efforts to protect our trade secrets, our competitors may discover our trade secrets, either through breach of these agreements,
independent development or publication of information including our trade secrets in cases where we do not have proprietary or otherwise
protected rights at the time of publication. A competitor’s discovery of our trade secrets would impair our competitive position
and have an adverse impact on our business.
Risks
Related to Our Intellectual Property
If
we are unable to obtain and maintain patent and other intellectual property protection for our products and product candidates, or if
the scope of the patent and other intellectual property protection obtained is not sufficiently broad, our competitors could develop
and commercialize products similar or identical to ours, and our ability to successfully commercialize our products and product candidates
may be adversely affected.
Our
ability to compete effectively will depend, in part, on our ability to maintain the proprietary nature of our technology and manufacturing
processes. We rely on research, manufacturing and other know-how, patents, trade secrets, license agreements and contractual provisions
to establish our intellectual property rights and protect our products and product candidates. These legal means, however, afford only
limited protection and may not adequately protect our rights. As of December 11, 2023, our intellectual property portfolio includes 19
total pending patent applications and issued patents, inclusive of 1 issued patent in the U.S., 1 Decision to Grant in each of Japan,
Russia and New Zealand, and 9 PCT applications within the 5007 patent family - also, 6 pending provisional applications covering formulations,
manufacturing processes and methods of use.
In
certain situations and as considered appropriate, we have sought, and we intend to continue to seek to protect our proprietary position
by filing patent applications in the United States and, in at least some cases, one or more countries outside the United States relating
to current and future products and product candidates that are important to our business. However, we cannot predict whether the patent
applications currently being pursued will issue as patents, or whether the claims of any resulting patents will provide us with a competitive
advantage or whether we will be able to successfully pursue patent applications in the future relating to our current or future products
and product candidates. Moreover, the patent application and approval process is expensive and time-consuming. We may not be able to
file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. Furthermore, we, or any
future partners, collaborators, or licensees, may fail to identify patentable aspects of inventions made in the course of development
and commercialization activities before it is too late to obtain patent protection on them. Therefore, we may miss potential opportunities
to seek additional patent protection. It is possible that defects of form in the preparation or filing of patent applications may exist,
or may arise in the future, for example with respect to proper priority claims, inventorship, claim scope, or requests for patent term
adjustments. If we fail to establish, maintain or protect such patents and other intellectual property rights, such rights may be reduced
or eliminated. If there are material defects in the form, preparation, prosecution or enforcement of our patents or patent applications,
such patents may be invalid and/or unenforceable, and such applications may never result in valid, enforceable patents.
Even
if they are unchallenged, our patents and patent applications, if issued, may not provide us with any meaningful protection or prevent
competitors from designing around our patent claims by developing similar or alternative technologies or therapeutics in a non-infringing
manner. For example, a third party may develop a competitive therapy that provides benefits similar to one or more of our product candidates
but that falls outside the scope of our patent protection. If the patent protection provided by the patents and patent applications we
hold or pursue with respect to our product candidates is not sufficiently broad to impede such competition, our ability to successfully
commercialize our product candidates could be negatively affected.
As
discussed under the heading “BUSINESS”, our PCT patent application having international patent application number PCT/US2018/00085
received an application filing date of February 20, 2018, which is four days after the one year anniversary of the filing date of U.S.
provisional patent applications U.S. 62/459,975 and U.S. 62/459,981 to which the PCT patent application claims a priority benefit due
to a computer issue at the PCT receiving office. Despite the restoration of the priority benefit to the filing date of U.S. provisional
patent applications (U.S. 62/459,975 and U.S. 62/459,981) by the PCT, some countries in which national stage patent applications were
filed from this PCT patent application did not accept this restoration including Canada and China, and the restoration procedure is pending
in Brazil. In the event that priority is not restored, prior art may be available to these patent applications that may otherwise not
be available to other patent applications filed from PCT/US2018/00085. This could affect the scope or breadth of the patent claims we
are pursuing in Brazil, Canada, and China, or could result in no ability to receive patents in these countries.
Other
parties, many of whom have substantially greater resources and have made significant investments in competing technologies, have developed
or may develop technologies that may be related or competitive with our approach, and may have filed or may file patent applications
and may have been issued or may be issued patents with claims that overlap or conflict with our patent applications, either by claiming
the same compositions, formulations or methods or by claiming subject matter that could dominate our patent position. In addition, the
laws of foreign countries may not protect our rights to the same extent as the laws of the United States. As a result, any patents we
may obtain in the future may not provide us with adequate and continuing patent protection sufficient to exclude others from commercializing
products similar to our products and product candidates.
The
patent position of biotechnology and pharmaceutical companies generally is highly uncertain. No consistent policy regarding the breadth
of claims allowed in biotechnology and pharmaceutical patents has emerged to date in the United States or in many foreign jurisdictions.
In addition, the determination of patent rights with respect to pharmaceutical compounds commonly involves complex legal and factual
questions, which has in recent years been the subject of much litigation. As a result, the issuance, scope, validity, enforceability
and commercial value of our patent rights are highly uncertain. Our competitors may also seek approval to market their own products similar
to or otherwise competitive with our products. Alternatively, our competitors may seek to market generic versions of any approved products
by submitting ANDAs to the FDA in which they claim that our patents are invalid, unenforceable or not infringed. In these circumstances,
we may need to defend or assert our patents, or both, including by filing lawsuits alleging patent infringement. In any of these types
of proceedings, a court or other agency with jurisdiction may find our patents invalid or unenforceable, or that our competitors are
competing in a non-infringing manner. Thus, even if we have valid and enforceable patents, these patents still may not provide protection
against competing products or processes sufficient to achieve our business objectives.
In
the future, one or more of our products and product candidates may be in-licensed from third parties. Accordingly, in some cases, the
availability and scope of potential patent protection is limited based on prior decisions by our licensors or the inventors, such as
decisions on when to file patent applications or whether to file patent applications at all. Our failure to obtain, maintain, enforce
or defend such intellectual property rights, for any reason, could allow third parties, in particular, other established and better financed
competitors having established development, manufacturing and distribution capabilities, to make competing products or impact our ability
to develop, manufacture and market our products and product candidates, even if approved, on a commercially viable basis, if at all,
which could have a material adverse effect on our business.
In
addition to patent protection, we expect to rely heavily on trade secrets, know-how and other unpatented technology, which are difficult
to protect. Although we seek such protection in part by entering into confidentiality agreements with our vendors, employees, consultants
and others who may have access to proprietary information, we cannot be certain that these agreements will not be breached, adequate
remedies for any breach would be available, or our trade secrets, know-how and other unpatented proprietary technology will not otherwise
become known to or be independently developed by our competitors. If we are unsuccessful in protecting our intellectual property rights,
sales of our products may suffer and our ability to generate revenue could be severely impacted.
Issued
patents covering our products and product candidates could be found invalid or unenforceable if challenged in court or in administrative
proceedings. We may not be able to protect our trade secrets in court.
If
we initiate legal proceedings against a third-party to enforce a patent covering one of our products or product candidates, should such
a patent issue, the defendant could counterclaim that the patent covering our product or product candidate is invalid or unenforceable.
In patent litigation in the United States, defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for
a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness,
written description or non- enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with
prosecution of the patent withheld information material to patentability from the USPTO, or made a misleading statement, during prosecution.
Third parties also may raise similar claims before administrative bodies in the United States or abroad, even outside the context of
litigation. Such mechanisms include re- examination, post grant review, inter partes review and equivalent proceedings in foreign jurisdictions.
An adverse determination in any of the foregoing proceedings could result in the revocation or cancellation of, or amendment to, our
patents in such a way that they no longer cover our products or product candidates. The outcome following legal assertions of invalidity
and unenforceability is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating
prior art, of which the patent examiner and we were unaware during prosecution. If a defendant or third party were to prevail on a legal
assertion of invalidity or unenforceability, we could lose at least part, and perhaps all, of the patent protection on one or more of
our products and product candidates. Such a loss of patent protection could have a material adverse impact on our business.
In
addition, our trade secrets may otherwise become known or be independently discovered by competitors. Competitors and other third parties
could purchase our products and product candidates and attempt to replicate some or all of the competitive advantages we derive from
our development efforts, willfully infringe, misappropriate or otherwise violate our intellectual property rights, design around our
protected technology or develop their own competitive technologies that fall outside of our intellectual property rights. If any of our
trade secrets were to be lawfully obtained or independently developed by a competitor or other third party, we would have no right to
prevent them, or those to whom they communicate it, from using that technology or information to compete with us. If our trade secrets
are not adequately protected or sufficient to provide an advantage over our competitors, our competitive position could be adversely
affected, as could our business. Additionally, if the steps taken to maintain our trade secrets are deemed inadequate, we may have insufficient
recourse against third parties for misappropriating our trade secrets.
We
may be subject to claims challenging the inventorship or ownership of the patents and other intellectual property.
We
may be subject to claims that former employees, collaborators or other third parties have an ownership interest in the patents and intellectual
property that we own or that we may own or license in the future. While it is our policy to require our employees and contractors who
may be involved in the development of intellectual property to execute agreements assigning such intellectual property to us, we may
be unsuccessful in executing such an agreement with each party who in fact develops intellectual property that we regard as our own or
such assignments may not be self-executing or may be breached. We could be subject to ownership disputes arising, for example, from conflicting
obligations of employees, consultants or others who are involved in developing our products or product candidates. Litigation may be
necessary to defend against any claims challenging inventorship or ownership. If we or fail in defending any such claims, we may have
to pay monetary damages and may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, intellectual
property, which could adversely impact our business, results of operations and financial condition.
Obtaining
and maintaining patent protection depends on compliance with various procedural, document submission, fee payment and other requirements
imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non- compliance with these requirements.
Periodic
maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and applications are required to be paid
to the USPTO and various governmental patent agencies outside of the United States in several stages over the lifetime of the patents
and applications. The USPTO and various non-U.S. governmental patent agencies require compliance with a number of procedural, documentary,
fee payment and other similar provisions during the patent application process and after a patent has issued. There are situations in
which non- compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of
patent rights in the relevant jurisdiction. The terms of one or more licenses that we enter into the future may not provide us with the
ability to maintain or prosecute patents in the portfolio, and must therefore rely on third parties to do so.
If
we do not obtain patent term extension and data exclusivity for our products and product candidates, our business may be materially harmed.
Patents
have a limited lifespan. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally
20 years from its earliest U.S. non-provisional filing date. Various extensions may be available, but the life of a patent, and the protection
it affords, is limited. Even if patents covering our product candidates are obtained, once the patent life has expired for a product
candidate, we may be open to competition from competitive products. Given the amount of time required for the development, testing and
regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates
are commercialized. As a result, our patent portfolio may not provide us with sufficient rights to exclude others from commercializing
products similar or identical to ours.
In
the future, if we obtain an issued patent covering one of our present or future product candidates, depending upon the timing, duration
and specifics of any FDA marketing approval of such product candidates, such patent may be eligible for limited patent term extension
under the Drug Price Competition and Patent Term Restoration Act of 1984, or Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit
a patent extension term of up to five years as compensation for patent term lost during the FDA regulatory review process. A patent term
extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent
may be extended and only those claims covering the approved drug, a method for using it or a method for manufacturing it may be extended.
A patent may only be extended once and only based on a single approved product. However, we may not be granted an extension because of,
for example, failure to obtain a granted patent before approval of a product candidate, failure to exercise due diligence during the
testing phase or regulatory review process, failure to apply within applicable deadlines, failure to apply prior to expiration of relevant
patents or otherwise our failure to satisfy applicable requirements. A patent licensed to us by a third party may not be available for
patent term extension. Moreover, the applicable time period or the scope of patent protection afforded could be less than we request.
If we are unable to obtain patent term extension or the term of any such extension is less than we request, our competitors may obtain
approval of competing products following our patent expiration, and our revenue could be reduced, possibly materially.
Changes
in patent law in the United States and other jurisdictions could diminish the value of patents in general, thereby impairing our ability
to protect our products and product candidates.
Changes
in either the patent laws or the interpretation of the patent laws in the United States or other jurisdictions could increase the uncertainties
and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents. On September 16, 2011,
the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. When implemented, the Leahy-Smith Act included several
significant changes to U.S. patent law that impacted how patent rights could be prosecuted, enforced and defended. In particular, the
Leahy-Smith Act also included provisions that switched the United States from a “first-to-invent” system to a “first-to-file”
system, allowed third- party submission of prior art to the USPTO during patent prosecution and set forth additional procedures to attack
the validity of a patent by the USPTO administered post grant proceedings. Under a first-to-file system, assuming the other requirements
for patentability are met, the first inventor to file a patent application generally will be entitled to the patent on an invention regardless
of whether another inventor had made the invention earlier. The USPTO developed new regulations and procedures governing the administration
of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the
first to file provisions, only became effective on March 16, 2013. It remains unclear what, if any, impact the Leahy-Smith Act will have
on the operation of our business. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding
the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse
effect on our business.
In
addition, the patent positions of companies in the development and commercialization of biologics and pharmaceuticals are particularly
uncertain. Recent rulings from the U.S. Court of Appeals for the Federal Circuit and the U.S. Supreme Court have narrowed the scope of
patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. This combination
of events has created uncertainty with respect to the validity and enforceability of patents, once obtained. Depending on future actions
by the U.S. Congress, the federal courts, and the USPTO, the laws and regulations governing patents could change in unpredictable ways
that could have a material adverse effect on our existing patent portfolio and our ability to protect and enforce our intellectual property
in the future.
We
cannot assure you that our efforts to seek patent protection for one or more of our products and product candidates will not be negatively
impacted by the decisions described above, rulings in other cases or changes in guidance or procedures issued by the USPTO. We cannot
fully predict what impact courts’ decisions in historical and future cases may have on the ability of life science companies to
obtain or enforce patents relating to their products in the future. These decisions, the guidance issued by the USPTO and rulings in
other cases or changes in USPTO guidance or procedures could have a material adverse effect on our existing patent rights and our ability
to protect and enforce our intellectual property in the future.
We
may not be able to protect our intellectual property rights throughout the world.
Filing,
prosecuting, maintaining, defending and enforcing patents on products and product candidates in all countries throughout the world would
be prohibitively expensive, and our intellectual property rights in some countries outside the United States could be less extensive
than those in the United States. The requirements for patentability may differ in certain countries, particularly in developing countries;
thus, even in countries where we do pursue patent protection, there can be no assurance that any patents will issue with claims that
cover our products. There can be no assurance that we will obtain or maintain patent rights in or outside the United States under any
future license agreements. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent
as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions
in all countries outside the United States, even in jurisdictions where we pursue patent protection, or from selling or importing products
made using our inventions in and into the United States or other jurisdictions. Competitors may use our technologies in jurisdictions
where we have not pursued and obtained patent protection to develop their own products and, further, may export otherwise infringing
products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These products
may compete with our products and product candidates and our patents or other intellectual property rights may not be effective or sufficient
to prevent them from competing.
Many
companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The
legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets
and other intellectual property protection, particularly those relating to biotechnology and pharmaceutical products, which could make
it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights
generally. For example, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third
parties. Proceedings to enforce our patent rights, even if obtained, in foreign jurisdictions could result in substantial costs and divert
our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly
and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in
any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. While we intend
to protect our intellectual property rights in major markets for our products, we cannot ensure that we will be able to initiate or maintain
similar efforts in all jurisdictions in which we may wish to market our products. Accordingly, our efforts to enforce our intellectual
property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we
develop.
If
we are sued for infringing intellectual property rights of third parties, such litigation could be costly and time consuming and could
prevent or delay us from developing or commercializing our product candidates.
Our
commercial success depends, in part, on our ability to develop, manufacture, market and sell our product candidates without infringing
the intellectual property and other proprietary rights of third parties. Third parties may have U.S. and non-U.S. issued patents and
pending patent applications relating to compounds, methods of manufacturing compounds and/or methods of use for the treatment of the
disease indications for which we are developing our product candidates. If any third-party patents or patent applications are found to
cover our product candidates or their methods of use or manufacture, we and our collaborators or sublicensees may not be free to manufacture
or market our product candidates as planned without obtaining a license, which may not be available on commercially reasonable terms,
or at all. We may also be required to indemnify our collaborators or sublicensees in such an event.
There
is a substantial amount of intellectual property litigation in the biotechnology and pharmaceutical industries, and we may become party
to, or threatened with, litigation or other adversarial proceedings regarding intellectual property rights with respect to our products
candidates, including interference and post-grant proceedings before the USPTO. There may be third-party patents or patent applications
with claims to materials, formulations, methods of manufacture or methods for treatment related to the composition, use or manufacture
of our product candidates. We cannot guarantee that any of our patent searches or analyses including, but not limited to, the identification
of relevant patents, the scope of patent claims or the expiration of relevant patents are complete or thorough, nor can we be certain
that we have identified each and every patent and pending application in the United States and abroad that is relevant to or necessary
for the commercialization of our product candidates in any jurisdiction. Because patent applications can take many years to issue, there
may be currently pending patent applications which may later result in issued patents that our product candidates may be accused of infringing.
In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. Accordingly,
third parties may assert infringement claims against us based intellectual property rights that exist now or arise in the future. The
outcome of intellectual property litigation is subject to uncertainties that cannot be adequately quantified in advance. The pharmaceutical
and biotechnology industries have produced a significant number of patents, and it may not always be clear to industry participants,
including us, which patents cover various types of products or methods of use or manufacture. The scope of protection afforded by a patent
is subject to interpretation by the courts, and the interpretation is not always uniform. If we were sued for patent infringement, we
would need to demonstrate that our product candidates, products or methods either do not infringe the patent claims of the relevant patent
or that the patent claims are invalid or unenforceable, and we may not be able to do this. Proving invalidity is difficult. For example,
in the United States, proving invalidity requires a showing of clear and convincing evidence to overcome the presumption of validity
enjoyed by issued patents. Even if we are successful in these proceedings, we may incur substantial costs and the time and attention
of our management and scientific personnel could be diverted in pursuing these proceedings, which could significantly harm our business
and operating results. In addition, we may not have sufficient resources to bring these actions to a successful conclusion.
If
we are found to infringe a third party’s intellectual property rights, we could be forced, including by court order, to cease developing,
manufacturing or commercializing the infringing product candidate or product. Alternatively, we may be required to obtain a license from
such third party in order to use the infringing technology and continue developing, manufacturing or marketing the infringing product
candidate or product.
However,
we may not be able to obtain any required license on commercially reasonable terms or at all. Even if we were able to obtain a license,
it could be non-exclusive, thereby giving our competitors access to the same technologies licensed to us; alternatively or additionally
it could include terms that impede or destroy our ability to compete successfully in the commercial marketplace. In addition, we could
be found liable for monetary damages, including treble damages and attorneys’ fees if we are found to have willfully infringed
a patent. A finding of infringement could prevent us from commercializing our product candidates or force us to cease some of our business
operations, which could harm our business. Claims that we have misappropriated the confidential information or trade secrets of third
parties could have a similar negative impact on our business.
We
may be subject to claims by third parties asserting that our employees or we have misappropriated their intellectual property, or claiming
ownership of what we regard as our own intellectual property.
Many
of our current and former employees, including our senior management, were previously employed at universities or at other biotechnology
or pharmaceutical companies, including some which may be competitors or potential competitors. Some of these employees may be subject
to proprietary rights, non-disclosure and non- competition agreements, or similar agreements, in connection with such previous employment.
Although we try to ensure that our employees do not use the proprietary information or know-how of others in their work for us, we may
be subject to claims that we or these employees have used or disclosed intellectual property, including trade secrets or other proprietary
information, of any such third party. Litigation may be necessary to defend against such claims. If we fail in defending any such claims,
in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel or sustain damages. Such intellectual
property rights could be awarded to a third party, and we could be required to obtain a license from such third party to commercialize
our technology or products. Such a license may not be available on commercially reasonable terms or at all. Even if we are successful
in defending against such claims, litigation could result in substantial costs and be a distraction to management.
In
addition, while we typically require our employees, consultants and contractors who may be involved in the development of intellectual
property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with
each party who in fact develops intellectual property that we regard as our own, which may result in claims by or against us related
to the ownership of such intellectual property. If we fail in prosecuting or defending any such claims, in addition to paying monetary
damages, we may lose valuable intellectual property rights. Even if we are successful in prosecuting or defending against such claims,
litigation could result in substantial costs and be a distraction to our senior management and scientific personnel.
We
may become involved in lawsuits to protect or enforce our patents and other intellectual property rights, which could be expensive, time-consuming
and unsuccessful.
Competitors
may infringe our patents, trademarks, copyrights or other intellectual property. To counter infringement or unauthorized use, we may
be required to file infringement claims, which can be expensive and time consuming and divert the time and attention of our management
and scientific personnel. In addition, our patents may become, involved in inventorship, priority, or validity disputes. To counter or
defend against such claims can be expensive and time-consuming, and our adversaries may have the ability to dedicate substantially greater
resources to prosecuting these legal actions than we can. Any claims we assert against perceived infringers could provoke these parties
to assert counterclaims against us alleging that we infringe their patents, in addition to counterclaims asserting that our patents are
invalid or unenforceable, or both.
In
an infringement proceeding, a court may decide that a patent is invalid or unenforceable, or may refuse to stop the other party from
using the technology at issue on the grounds that our patents do not cover the technology in question. Accordingly, despite our efforts,
we may not be able to prevent third parties from infringing upon or misappropriating intellectual property rights we own or control.
An adverse result in any litigation proceeding could put one or more of our owned or in-licensed patents at risk of being invalidated
or interpreted narrowly. Further, because of the substantial amount of discovery required in connection with intellectual property litigation,
there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation.
Even
if resolved in our favor, the court may decide not to grant an injunction against further infringing activity and instead award only
monetary damages, which may or may not be an adequate remedy. Litigation or other legal proceedings relating to intellectual property
claims may cause us to incur significant expenses and could distract our personnel from their normal responsibilities. In addition, there
could be public announcements of the results of hearings, motions, or other interim proceedings or developments, and if securities analysts
or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock. Such
litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities
or any future sales, marketing, or distribution activities.
We
may not have sufficient financial or other resources to conduct such litigation or proceedings adequately. Some of our competitors may
be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources
and more mature and developed intellectual property portfolios. Uncertainties resulting from the initiation and continuation of patent
litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace.
If
we fail to comply with our obligations under any future intellectual property licenses with third parties, we could lose license rights
that are important to our business.
In
connection with our efforts to build our product candidate pipeline, we may enter into license agreements in the future. We expect that
such license agreements will impose, various diligence, milestone payment, royalty, insurance and other obligations on us. If we fail
to comply with our obligations under these licenses, our licensors may have the right to terminate these license agreements, in which
event we might not be able to market any product that is covered by these agreements, or our licensors may convert the license to a non-exclusive
license, which could negatively impact the value of the product candidate being developed under the license agreement. Termination of
these license agreements or reduction or elimination of our licensed rights may also result in our having to negotiate new or reinstated
licenses with less favorable terms.
If
our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our marks of interest
and our business may be adversely affected.
Our
trademarks or trade names may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks.
We rely on both registration and common law protection for our trademarks. We may not be able to protect our rights to these trademarks
and trade names or may be forced to stop using these names, which we need for name recognition by potential partners or customers in
our markets of interest. During trademark registration proceedings, we may receive rejections. Although we would be given an opportunity
to respond to those rejections, we may be unable to overcome such rejections. In addition, in the USPTO and in comparable agencies in
many foreign jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered
trademarks. Opposition or cancellation proceedings may be filed against our trademarks, and our trademarks may not survive such proceedings.
If we are unable to establish name recognition based on our trademarks and trade names, we may not be able to compete effectively and
our business may be adversely affected.
Risks
Related to Employee Matters and Managing Growth
We
only have a limited number of employees to manage and operate our business.
As
of September 30, 2023, we had about 12 full-time employees. Additionally, we utilize independent contractors and other third parties
to assist with various aspects of our business. Our focus on the development of our product candidates requires us to optimize cash utilization
and to manage and operate our business in a highly efficient manner. We cannot assure you that we will be able to hire or retain adequate
staffing levels to develop our product candidates or run our operations or to accomplish all of the objectives that we otherwise would
seek to accomplish.
Cyber-attacks
or other failures in telecommunications or information technology systems could result in information theft, data corruption and significant
disruption of our business operations.
We
utilize information technology, or IT, systems and networks to process, transmit and store electronic information in connection with
our business activities. As use of digital technologies has increased, cyber incidents, including deliberate attacks and attempts to
gain unauthorized access to computer systems and networks, have increased in frequency and sophistication. These threats pose a risk
to the security of our systems and networks, the confidentiality and the availability and integrity of our data.
Our
future success depends on our ability to retain key employees, consultants and advisors and to attract, retain and motivate qualified
personnel.
We
are highly dependent on principal members of our executive team and key employees, the loss of whose services may adversely impact the
achievement of our objectives. While we have entered into employment agreements with certain of our executive officers, any of them could
leave our employment at any time. We do not maintain “key person” insurance policies on the lives of these individuals or
the lives of any of our other employees. The loss of the services of one or more of our current employees might impede the achievement
of our research, development and commercialization objectives. Furthermore, replacing executive officers or other key employees may be
difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills
and experience required to develop, gain marketing approval of and commercialize products successfully.
Recruiting
and retaining other qualified employees, consultants and advisors for our business, including scientific and technical personnel, will
also be critical to our success. There is currently a shortage of skilled executives in our industry, which is likely to continue. As
a result, competition for skilled personnel is intense and the turnover rate can be high. We may not be able to attract and retain personnel
on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for individuals with similar skill
sets. In addition, failure to succeed in preclinical or clinical trials may make it more challenging to recruit and retain qualified
personnel.
In
addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and
development and commercialization strategy. Our consultants and advisors may be employed by other entities and may have commitments under
consulting or advisory contracts with those entities that may limit their availability to us. If we are unable to continue to attract
and retain highly qualified personnel, our ability to develop and commercialize our product candidates will be limited.
The
inability to recruit or the loss of the services of any executive, key employee, consultant or advisor may impede the progress of our
research, development and commercialization objectives.
Our
employees, independent contractors, consultants, collaborators and contract research organizations may engage in misconduct or other
improper activities, including non-compliance with regulatory standards and requirements, which could cause significant liability for
us and harm our reputation.
We
are exposed to the risk that our employees, independent contractors, consultants, collaborators and contract research organizations may
engage in fraudulent conduct or other illegal activity. Misconduct by those parties could include intentional, reckless and/or negligent
conduct or disclosure of unauthorized activities to us that violates: (1) FDA regulations or similar regulations of comparable non-U.S.
regulatory authorities, including those laws requiring the reporting of true, complete and accurate information to such authorities,
(2) manufacturing standards, (3) federal and state healthcare fraud and abuse laws and regulations and similar laws and regulations established
and enforced by comparable non-U.S. regulatory authorities, and (4) laws that require the reporting of financial information or data
accurately. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations
intended to prevent fraud, misconduct, kickbacks, self- dealing, bribery and other abusive practices. These laws and regulations restrict
or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business
arrangements. Employee or collaborator misconduct could also involve the improper use of, including trading on, information obtained
in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. While we have a code
of conduct and business ethics, it is not always possible to identify and deter misconduct, and the precautions we take to detect and
prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental
investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws, standards or regulations. If
any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could
have a significant impact on our business and results of operations, including the imposition of civil, criminal and administrative penalties,
damages, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, additional
reporting requirements and/or oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations
of non-compliance with these laws, imprisonment, contractual damages, reputational harm, diminished profits and future earnings, and
curtailment of our operations, any of which could have a material adverse effect on our ability to operate our business and our results
of operations.
We
expect to expand our organization, and as a result, we may encounter difficulties in managing our growth, which could disrupt our operations.
We
expect to experience significant growth in the number of our employees and the scope of our operations, particularly in the areas of
drug manufacturing, regulatory affairs and sales, marketing and distribution, as well as to support our public company operations. To
manage these growth activities, we must continue to implement and improve our managerial, operational and financial systems, expand our
facilities and continue to recruit and train additional qualified personnel. Our management may need to devote a significant amount of
its attention to managing these growth activities. Moreover, our expected growth could require us to relocate to geographic areas beyond
those where we have been historically located. For example, we maintain an office in Princeton, New Jersey, at which many of our finance,
management and administrative personnel work. Due to our limited financial resources and the limited experience of our management team
in managing a company with such anticipated growth, we may not be able to effectively manage the expansion or relocation of our operations,
retain key employees, or identify, recruit and train additional qualified personnel. Our inability to manage the expansion or relocation
of our operations effectively may result in weaknesses in our infrastructure, give rise to operational mistakes, loss of business opportunities,
loss of employees and reduced productivity among remaining employees. Our expected growth could also require significant capital expenditures
and may divert financial resources from other projects, such as the development of additional product candidates. If we are unable to
effectively manage our expected growth, our expenses may increase more than expected, our ability to generate revenues could be reduced
and we may not be able to implement our business strategy, including the successful commercialization of our product candidates.
Risks
Related to Our Common Stock
The
market price of our common stock may be significantly volatile.
The
market price for our common stock may be volatile and subject to wide fluctuations in response to factors including the following:
| ● |
actual
or anticipated fluctuations in our quarterly or annual operating results; |
| |
|
| ● |
changes
in financial or operational estimates or projections; |
| |
|
| ● |
conditions
in markets generally; |
| |
|
| ● |
changes
in the economic performance or market valuations of companies similar to ours; and |
| |
|
| ● |
general
economic or political conditions in the United States or elsewhere. |
In
particular, the market prices of biotechnology companies like ours have been highly volatile due to factors, including, but not limited
to:
| ● |
any
delay or failure to conduct a clinical trial for our product or receive approval from the FDA and other regulatory agencies; |
| |
|
| ● |
developments
or disputes concerning a company’s intellectual property rights; |
| |
|
| ● |
technological
innovations of such companies or their competitors; |
| |
|
| ● |
changes
in market valuations of similar companies; |
| |
|
| ● |
announcements
by such companies or their competitors of significant contracts, acquisitions, strategic partnerships, joint ventures, capital commitments,
new technologies, or patents; and |
| |
|
| ● |
failure
to complete significant transactions or collaborate with vendors in manufacturing a product. |
The
securities market has from time to time experienced significant price and volume fluctuations that are not related to the operating performance
of particular companies. These market fluctuations may also materially and adversely affect the market price of shares of our common
stock.
We
do not expect to pay cash dividends in the foreseeable future and therefore investors should not anticipate cash dividends on their investment.
Our
board of directors does not intend to pay cash dividends in the foreseeable future but instead intends to retain any and all earnings
to finance the growth of the business. To date, we have not paid any cash dividends and there can be no assurance that cash dividends
will ever be paid on our common stock.
We
incur significant costs and devote substantial management time as a result of operating as a public company, and we expect those costs
to increase.
As
a public company, we incur significant legal, accounting and other expenses. For example, we are required to comply with certain of the
requirements of the Sarbanes-Oxley Act and the Dodd-Frank Wall Street Reform and Consumer Protection Act, as well as rules and regulations
subsequently implemented by the SEC, including the establishment and maintenance of effective disclosure and financial controls and changes
in corporate governance practices. We expect that compliance with these requirements will increase our legal and financial compliance
costs and will make some activities more time consuming and costly. In addition, we expect that our management and other personnel will
need to divert attention from operational and other business matters to devote substantial time to these public company requirements.
In particular, we expect to incur significant expenses and devote substantial management effort toward ensuring compliance with the requirements
of Section 404 of the Sarbanes-Oxley Act. We currently do not have an internal audit function, and we may need to hire or contract for
additional accounting and financial staff with appropriate public company experience and technical accounting knowledge.
There
may be limitations on the effectiveness of our internal controls, and a failure of our control systems to prevent error or fraud may
materially harm our company.
We
are required, pursuant to Section 404 of the Sarbanes-Oxley Act, to furnish a report by our management on, among other things, the effectiveness
of our internal control over financial reporting. This assessment will need to include disclosure of any material weaknesses identified
by our management in our internal control over financial reporting. A material weakness is a deficiency, or combination of deficiencies,
in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of annual or interim
consolidated financial statements will not be prevented or detected on a timely basis.
Effective
internal control over financial reporting is necessary for us to provide reliable and timely financial reports and, together with adequate
disclosure controls and procedures, are designed to reasonably detect and prevent fraud. Any failure to implement required new or improved
controls, or difficulties encountered in their implementation could cause us to fail to meet our reporting obligations. Undetected material
weaknesses in our internal control over financial reporting could lead to financial statement restatements and require us to incur the
expense of remediation.
Moreover,
we do not expect that disclosure controls or internal control over financial reporting will prevent all error and all fraud. A control
system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s
objectives will be met. Further, the design of a control system must reflect the fact that there are resource constraints and the benefits
of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of
controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected. Failure of our control
systems to detect or prevent error or fraud could materially adversely impact us.
Any
of the foregoing occurrences, should they come to pass, could negatively impact the public perception of our company, which could have
a negative impact on our stock price.
We
may be unable to complete our analysis of our internal controls over financial reporting in a timely manner, or these internal controls
may not be determined to be effective, which may adversely affect investor confidence in our company and, as a result, the value of our
common stock.
We
may not be able to complete our evaluation and testing of our internal control over financial reporting. During the evaluation and testing
process, if we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert
that our internal controls are effective.
If
we are unable to assert that our internal control over financial reporting is effective, or, if applicable, our independent registered
public accounting firm is unable to express an opinion on the effectiveness of our internal controls, we could lose investor confidence
in the accuracy and completeness of our financial reports, which would cause the price of our common stock to decline, and we may be
subject to investigation or sanctions by the SEC. We will also be required to disclose changes made in our internal control and procedures
on a quarterly basis.
Anti-takeover
provisions under Delaware law could make an acquisition of the combined company more difficult and may prevent attempts by the combined
company stockholders to replace or remove the combined company management.
Because
the combined company will be incorporated in Delaware, it is governed by the provisions of Section 203 of the Delaware General Corporation
Law, or the DGCL, which prohibits stockholders owning in excess of 15% of the outstanding combined company voting stock from merging
or combining with the combined company. Although we believe these provisions collectively will provide for an opportunity to receive
higher bids by requiring potential acquirers to negotiate with the combined company’s board of directors, they would apply even
if the offer may be considered beneficial by some stockholders. In addition, these provisions may frustrate or prevent any attempts by
the combined company’s stockholders to replace or remove then current management by making it more difficult for stockholders to
replace members of the board of directors, which is responsible for appointing the members of management.
Director
and officer liability is limited.
As
permitted by Delaware law, our bylaws limit the liability of our directors for monetary damages for breach of a director’s fiduciary
duty except for liability in certain instances. As a result of our bylaw provisions and Delaware law, stockholders may have limited rights
to recover against directors for breach of fiduciary duty.
General
Risk Factors
Cyber-attacks
or other failures in telecommunications or information technology systems could result in information theft, data corruption and significant
disruption of our business operations.
We
utilize information technology, or IT, systems and networks to process, transmit and store electronic information in connection with
our business activities. As use of digital technologies has increased, cyber incidents, including deliberate attacks and attempts to
gain unauthorized access to computer systems and networks, have increased in frequency and sophistication. These threats pose a risk
to the security of our systems and networks, the confidentiality and the availability and integrity of our data.
Our
common stock could be further diluted as the result of the issuance of additional shares of common stock, convertible securities, warrants
or options.
In
the past, we have issued common stock, convertible securities (such as convertible notes) and warrants in order to raise capital. We
have also issued common stock as compensation for services and incentive compensation for our employees, directors and certain vendors.
We have shares of common stock reserved for issuance upon the exercise of certain of these securities and may increase the shares reserved
for these purposes in the future. Our issuance of additional common stock, convertible securities, options and warrants could affect
the rights of our stockholders, could reduce the market price of our common stock or could result in adjustments to exercise prices of
outstanding warrants (resulting in these securities becoming exercisable for, as the case may be, a greater number of shares of our common
stock), or could obligate us to issue additional shares of common stock to certain of our stockholders.
Shares
eligible for future sale may adversely affect the market.
From
time to time, certain of our stockholders may be eligible to sell all or some of their shares of common stock by means of ordinary brokerage
transactions in the open market pursuant to Rule 144 promulgated under the Securities Act, subject to certain limitations. In general,
pursuant to Rule 144, stockholders who have been non-affiliates for the preceding three months may sell shares of our common stock freely
after six months subject only to the current public information requirement. Affiliates may sell shares of our common stock after six
months subject to the Rule 144 volume, manner of sale, current public information and notice requirements. Any substantial sales of our
common stock pursuant to Rule 144 may have a material adverse effect on the market price of our common stock.
Item
1B. Unresolved Staff Comments.
Not
applicable.
Item
2. Properties.
The
Company relies on short term office use contracts to procure office and meeting space.
Item
3. Legal Proceedings.
We
are not currently subject to any material legal proceedings. However, we may from time to time become a party to various legal proceedings
arising in the ordinary course of our business.
Item
4. Mine Safety Disclosures.
Not
applicable.
PART
II
Item
5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market
Information
The
Company’s common stock trades on The Nasdaq Capital Market under the symbol “SONN.”
Holders
As
of December 11, 2023, we had 76 holders of record of our common stock. The number of record holders was determined from the records
of the transfer agent and does not include beneficial owners of common stock whose shares are held in the names of various security brokers,
dealers, and registered clearing agencies. The transfer agent of our common stock is Securities Transfer Corporation, 2901 N Dallas Parkway,
Suite 380, Plano, TX 75093.
Dividends
We
have never declared or paid cash dividends on our common stock. We do not intend to declare or pay cash dividends on our common stock
for the foreseeable future, but currently intend to retain any future earnings to fund the development and growth of our business. The
payment of cash dividends if any, on the common stock will rest solely within the discretion of our board of directors and will depend,
among other things, upon our earnings, capital requirements, financial condition, and other relevant factors.
Item
6. [Reserved]
Item
7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
The
following Management’s Discussion and Analysis of Financial Condition and Results of Operations (“MD&A”) is intended
to help facilitate an understanding of our financial condition and our historical results of operations for the periods presented. This
MD&A should be read in conjunction with the audited consolidated financial statements and notes thereto included in this Annual Report
on Form 10-K. This MD&A may contain forward-looking statements that involve risks and uncertainties. For a discussion on forward-looking
statements, see the information set forth above under the caption “Special Note Regarding Forward-Looking Statements,” which
information is incorporated herein by reference.
Overview
Sonnet
BioTherapeutics Holdings, Inc. (“Sonnet,” “we,” “us,” “our” or the “Company”),
is a clinical stage, oncology-focused biotechnology company with a proprietary platform for innovating biologic medicines of single or bifunctional action. Known as FHAB™ (Fully Human Albumin Binding), the technology utilizes a fully human single chain
antibody fragment that binds to and “hitch-hikes” on human serum albumin for transport to target tissues. We designed the
construct to improve drug accumulation in specific tissues, as well as to extend the duration of activity in the body. FHAB
development candidates are produced in a mammalian cell culture, which enables glycosylation, thereby reducing the risk of immunogenicity.
We believe our FHAB technology, for which we received a U.S. patent in June 2021, is a distinguishing feature of our biopharmaceutical
platform that is well suited for future drug development across a range of human disease areas, including in oncology, autoimmune, pathogenic,
inflammatory, and hematological conditions.
Our
current internal pipeline development activities are focused on cytokines, a class of cell signaling peptides that, among other important
functions, serve as potent immunomodulatory agents. Working both independently and synergistically, specific cytokines have shown the
ability to modulate the activation and maturation of immune cells that fight cancer and pathogens. However, because they do not preferentially
accumulate in specific tissues and are quickly eliminated from the body, the conventional approach to achieving a treatment effect with
cytokine therapy typically requires the administration of high and frequent doses. This can result in a reduced treatment effect accompanied
by the potential for systemic toxicity, which poses challenges to the therapeutic application of this class of drugs.
Our
lead proprietary asset, SON-1010, is a fully human version of Interleukin 12 (“IL-12”), covalently linked to the FHAB
construct, for which we are pursuing clinical development in solid tumor indications, including ovarian cancer, non-small cell lung
cancer and head and neck cancer. In March 2022, the FDA cleared our Investigational New Drug (“IND”) application for SON-1010.
This allowed us to initiate a U.S. clinical trial (SB101) in oncology patients with solid tumors during the second calendar quarter of
2022. In September 2021, we created a wholly-owned Australian subsidiary, SonnetBio Pty Ltd (“Subsidiary”), for the purpose
of conducting certain clinical trials. We received approval and initiated an Australian clinical study (SB102) of SON-1010 in healthy
volunteers during the third calendar quarter of 2022. Interim safety and tolerability data from the SB101 and SB102 studies were reported
in April 2023.
In
January 2023, we announced a collaboration agreement with Roche for the clinical evaluation of SON-1010 with atezolizumab (Tecentriq®).
The companies have entered into a Master Clinical Trial and Supply Agreement (“MCSA”), along with ancillary Quality and Safety
Agreements, to study the safety and efficacy of the combination of SON-1010 and atezolizumab in a platinum-resistant ovarian cancer (“PROC”)
patient setting. Further, the companies will provide SON-1010 and atezolizumab, respectively, for use in the Phase 1b/Phase 2a combination
safety, dose-escalation, and efficacy study (SB221). Part 1 of this 2-part study was approved in June 2023 by the local Human Research Ethics Committee in Australia
under CT-2023-CTN-01399-1 and the Therapeutic Goods Administration has been notified. In August 2023, the FDA accepted the IND for SB221.
The trial consists of a modified 3+3 dose-escalation design in Part 1 to establish the maximum tolerated dose (MTD) of SON-1010 with a
fixed dose of atezolizumab. Clinical benefit in PROC will be confirmed in an expansion group to establish the recommended Phase 2 dose
(RP2D). Part 2 of the study will then investigate SON-1010 monotherapy, its use in combination with atezolizumab, or the standard of care
(SOC) for PROC in a randomized comparison to show proof-of-concept (POC).
We
acquired the global development rights to our most advanced compound, SON-080, a fully human version of Interleukin 6
(“IL-6”), in April 2020 through our acquisition of the outstanding shares of Relief Therapeutics SA. We are advancing
SON-080 in target indications of Chemotherapy-Induced Peripheral Neuropathy (“CIPN”) and Diabetic Peripheral Neuropathy
(“DPN”). We received approval to initiate an ex-U.S. Phase 1b/2a study with SON-080 in CIPN. Enrollment of the first
portion of the SB211 study in chemotherapy-induced peripheral neuropathy (CIPN) is nearing completion, which should position the
DSMB to complete its review of the preliminary safety data during the first calendar quarter of 2024. Pursuant to a license
agreement we entered into with New Life Therapeutics Pte, Ltd. (“New Life”) of Singapore in May 2021, Sonnet and New
Life will be jointly responsible for developing SON-080 in DPN. The objective will be to analyze the data and to consider initiating
a Phase 2 study, once the CIPN safety data has been evaluated.
SON-1210
(IL12-FHAB-IL15), our lead bi-specific construct, combines FHAB with fully human IL-12 and fully human Interleukin
15 (“IL-15”). This compound is being developed for solid tumor indications, including colorectal cancer. In February 2023,
we announced the successful completion of two IND-enabling toxicology studies with SON-1210 in non-human primates. We are prepared to
initiate the regulatory authorization process for SON-1210, pending the outcome of any partnering activity.
SON-1410
(IL18-FHAB-IL12) is a bi-specific combination of Interleukin 18 (“IL-18”) and IL-12 for solid tumor cancers. Cell line development and process development are ongoing, with early experimental
drug supply suitable for formulation and analytical method development activities. After some delays in 2023, activities will continue
into 2024 with the potential to generate a drug suitable for preclinical studies and subsequent human studies.
We
have completed sequence confirmation for SON-3015 (anti-IL6-FHAB-anti-TGFβ). Early stage bi-specific drug has been generated
and is being stored for future use in in vivo mice studies. We have elected to place the SON-3015 development program on hold
for expense reduction purposes.
As
part of the ongoing cost-cutting evaluations, all antiviral development with SON-1010 has been suspended.
We
have incurred recurring operating losses and negative cash flows since inception. Our ability to generate product or licensing revenue
sufficient to achieve profitability will depend heavily on the successful development and eventual commercialization of one or more of
our current or future product candidates. Our net losses were $18.8 million and $29.7 million for the years ended September 30, 2023
and 2022, respectively. As of September 30, 2023, we had cash of $2.3 million. We expect to continue to incur significant expenses and
increasing operating losses for at least the next several years. We expect that our expenses and capital requirements will increase substantially
in connection with our ongoing activities, particularly if and as we:
| ● | conduct
additional clinical trials for product candidates; |
| | | |
| ● | continue
to discover and develop additional product candidates; |
| | | |
| ● | acquire
or in-license other product candidates and technologies; |
| | | |
| ● | maintain,
expand and protect our intellectual property portfolio; |
| | | |
| ● | hire
additional clinical, scientific and commercial personnel; |
| ● | establish
a commercial manufacturing source and secure supply chain capacity sufficient to provide
commercial quantities of any product candidates for which we may obtain regulatory approval; |
| | | |
| ● | seek
regulatory approval for product candidates that successfully complete clinical trials; |
| | | |
| ● | establish
a sales, marketing and distribution infrastructure to commercialize any products for which
we may obtain regulatory approval; and |
| | | |
| ● | add
operational, financial and management information systems and personnel, including personnel
to support our product development and planned future commercialization efforts, as well
as to support our operation as a public reporting company. |
We
will not generate revenue from product sales, if any, unless and until we receive licensing revenue and/or successfully complete clinical
development and obtain regulatory approval for our product candidates. If we obtain regulatory approval for any of our product candidates
and do not enter into a commercialization partnership, we expect to incur significant expenses related to developing our internal commercialization
capability to support product sales, marketing and distribution. We will continue to incur significant costs associated with operating
as a public company.
As
a result, we will need substantial additional funding to support our continuing operations and pursue our growth strategy. Until such
time as we can generate significant revenue from product sales, if ever, we expect to finance our operations through the sale of equity,
debt financings or other capital sources, which may include collaborations with other companies or other strategic transactions. We may
not be able to raise additional funds or enter into such other agreements or arrangements when needed on favorable terms, or at all.
If we fail to raise capital or enter into such agreements as and when needed, we may have to significantly delay, reduce or eliminate
the development and commercialization of one or more of our product candidates or delay our pursuit of potential in-licenses or acquisitions.
Because
of the numerous risks and uncertainties associated with product development, we are unable to predict the timing or amount of increased
expenses or when or if we will be able to achieve or maintain profitability. Even if we are able to generate product sales, we may not
become profitable. If we fail to become profitable or are unable to sustain profitability on a continuing basis or raise additional capital
or enter into collaboration or license agreements, then we may be unable to continue our operations at planned levels and be forced to
reduce or terminate operations.
Since
our inception in 2015, we have devoted substantially all of our efforts and financial resources to organizing and staffing the Company,
business planning, raising capital, acquiring or discovering product candidates and securing related intellectual property rights and
conducting discovery, research and development activities for product candidates. We do not have any products approved for sale and have
not generated any revenue from product sales. We have funded our operations to date primarily with proceeds from sales of common stock,
warrants and proceeds from the issuance of convertible debt.
Components
of Results of Operations
Collaboration
Revenue
Collaboration
revenue is currently earned from the license arrangement entered into with New Life in May 2021, which granted New Life rights to an
exclusive license (with the right to sublicense) to develop and commercialize pharmaceutical preparations containing a specific recombinant
human IL-6, SON-080 (the “Compound”) (such preparations, the “Products”) for the prevention, treatment or palliation
of diabetic peripheral neuropathy in humans (the “DPN Field”) in the Exclusive Territory. We identified the following obligations
under the arrangement: (i) License to develop, market, import, use and commercialize the Product in the Field in the Exclusive Territory
(the “License”); and (ii) transfer of know-how and clinical development and regulatory activities (“R&D Activities”).
We determined that the License and the R&D Activities are not distinct from each other and, therefore, combined these material promises
into a single performance obligation. Under this agreement, we received upfront cash payments totaling $1.0 million, which were fully
allocated to the single performance obligation and are being recognized over the estimated performance period of R&D services.
Operating
Expenses
Research
and Development Expenses
Research
and development expenses consist primarily of costs incurred in connection with the discovery and development of our product candidates.
We expense research and development costs as incurred and such costs include:
| ● | employee-related
expenses, including salaries, share-based compensation and related benefits, for employees
engaged in research and development functions; |
| | | |
| ● | expenses
incurred in connection with the preclinical and clinical development of our product candidates,
including under agreements with third parties, such as consultants and clinical research
organizations; |
| | | |
| ● | the
cost of manufacturing drug products for use in our preclinical studies and clinical trials,
including under agreements with third parties, such as consultants and contract manufacturing
organizations; |
| | | |
| ● | facilities,
depreciation and other expenses, which include direct or allocated expenses for rent and
maintenance of facilities and insurance; |
| | | |
| ● | costs
related to compliance with regulatory requirements; and |
| | | |
| ● | payments
made under third-party licensing agreements. |
We
recognize external development costs based on an evaluation of the progress to completion of specific tasks using information provided
by our service providers. This process involves reviewing open contracts and purchase orders, communicating with their personnel to identify
services that have been performed on our behalf, and estimating the level of service performed and the associated cost incurred for the
service when we have not yet been invoiced or otherwise notified of actual costs. Nonrefundable advance payments for goods or services
to be received in the future for use in research and development activities are recorded as prepaid expenses. Such amounts are recognized
as an expense when the goods have been delivered or the services have been performed.
Our
direct research and development expenses consist primarily of external costs, such as fees paid to outside consultants, CROs, CMOs and
research laboratories in connection with preclinical development, process development, manufacturing and clinical development activities.
Our direct research and development expenses also include fees incurred under third-party license agreements. We do not allocate employee
costs and costs associated with discovery efforts, laboratory supplies and facilities, including depreciation or other indirect costs,
to specific product candidates because these costs are deployed across multiple programs and as such, are not separately classified.
We use internal resources primarily to conduct our research and discovery as well as for managing preclinical development, process development,
manufacturing and clinical development activities. These employees work across multiple programs and therefore, we do not track costs
by product candidate.
We
will continue to incur research and development expenses for the foreseeable future as we attempt to advance development of our product
candidates. The successful development of our product candidates is highly uncertain. At this time, we cannot reasonably estimate or
know the nature, timing and costs of the efforts that will be necessary to complete the remainder of the development of our current pipeline
or any future product candidates we may develop due to the numerous risks and uncertainties associated with clinical development, including
risk and uncertainties related to:
| ● | the
timing and progress of preclinical and clinical development activities; |
| | | |
| ● | the
number and scope of preclinical and clinical programs that we decide to pursue; |
| | | |
| ● | our
ability to maintain our current research and development programs and to establish new ones; |
| | | |
| ● | establishing
an appropriate safety profile with investigational new drug-enabling studies; |
| | | |
| ● | successful
patient enrollment in, and the initiation and completion of, clinical trials; |
| | | |
| ● | the
successful completion of clinical trials with safety, tolerability and efficacy profiles
that are satisfactory to the FDA or any comparable foreign regulatory authority; |
| | | |
| ● | the
receipt of regulatory approvals from applicable regulatory authorities; |
| | | |
| ● | the
timing, receipt and terms of any marketing approvals from applicable regulatory authorities; |
| | | |
| ● | our
ability to establish new licensing or collaboration arrangements; |
| | | |
| ● | establishing
agreements with third-party manufacturers for clinical supply for our clinical trials and
commercial manufacturing, if any of our product candidates is approved; |
| | | |
| ● | development
and timely delivery of clinical-grade and commercial-grade drug formulations that can be
used in our clinical trials and for commercial launch; |
| | | |
| ● | obtaining,
maintaining, defending and enforcing patent claims and other intellectual property rights; |
| | | |
| ● | launching
commercial sales of product candidates, if approved, whether alone or in collaboration with
others; |
| | | |
| ● | maintaining
a continued acceptable safety profile of the product candidates following approval; and |
| | | |
| ● | the
potential impact of COVID-19 on operations which may affect among other things, the timing
of clinical trials, availability of raw materials, and the ability to access and secure testing
facilities. |
A
change in the outcome of any of these variables with respect to the development of our product candidates could significantly change
the costs and timing associated with the development of that product candidate. We may never succeed in obtaining regulatory approval
for any of our product candidates.
General
and Administrative Expenses
General
and administrative expenses consist primarily of salaries and related costs for personnel, including share-based compensation, in executive,
finance and administrative functions. General and administrative expenses also include direct and allocated facility-related costs as
well as professional fees for legal, patent, consulting, accounting, and audit services.
Our
general and administrative expenses will increase in the future as we increase our headcount to support continued research activities
and development of product candidates. We will continue to incur increased accounting, audit, legal, regulatory, compliance and director
and officer insurance costs as well as investor and public relations expenses associated with being a public company.
Foreign
Exchange Loss
Foreign
exchange loss consists of exchange rate changes on transactions denominated in currencies other than the U.S. dollar.
Results
of Operations
Comparison
of the Years Ended September 30, 2023 and 2022
The
following table summarizes our results of operations for the years ended September 30, 2023 and 2022:
| | |
Years ended September 30, | | |
| |
| | |
2023 | | |
2022 | | |
Change | |
| Collaboration revenue | |
$ | 147,805 | | |
$ | 349,943 | | |
$ | (202,138 | ) |
| Operating expenses: | |
| | | |
| | | |
| | |
| Research and development | |
| 11,814,690 | | |
| 21,444,019 | | |
$ | (9,629,329 | ) |
| General and administrative | |
| 7,125,732 | | |
| 8,575,283 | | |
| (1,449,551 | ) |
| Total operating expense | |
| 18,940,422 | | |
| 30,019,302 | | |
| (11,078,880 | ) |
| Loss from operations | |
| (18,8792,617 | ) | |
| (29,669,359 | ) | |
| 10,876,742 | |
| Foreign exchange loss | |
| (40,077 | ) | |
| (52,482 | ) | |
| 12,405 | |
| Net loss | |
$ | (18,832,694 | ) | |
$ | (29,721,841 | ) | |
$ | 10,889,147 | |
Collaboration
Revenue
We
recognized $0.1 million of revenue related to the New Life Agreement during the year ended September 30, 2023 compared to $0.3 million
during the year ended September 30, 2022. The decrease of $0.2 million was due to a delay in timing in the performance of R&D services.
Research
and Development Expenses
Research
and development expenses were $11.8 million for the year ended September 30, 2023, compared to $21.4 million for the year ended September
30, 2022. The decrease of $9.6 million was primarily due to the establishment of cost savings by transitioning product development activities
to cost advantaged locations such as India and Australia, by reducing expenditures on tertiary programs such as SON-3015, which has been
placed on a development hold, and suspending antiviral development related to SON-1010, as well as a decrease in share-based compensation
expense.
General
and Administrative Expenses
General
and administrative expenses were $7.1 million for the year ended September 30, 2023, compared to $8.6 million for the year ended September
30, 2022. The decrease of $1.5 million relates primarily to a decrease in share-based compensation, legal and business development expenses,
as we are managing expenses for liquidity purposes and are tightening our focus on the research and development projects we have assessed
to have the greatest near-term potential.
Liquidity
and Capital Resources
We
have funded operations to date primarily with proceeds from sales of common stock, warrants and proceeds from the issuance of convertible
debt. We will likely offer additional securities for sale in response to market conditions or other circumstances if we believe such
a plan of financing is required to advance our business plans and is in the best interests of our stockholders. There is no certainty
that equity or debt financing will be available in the future or that it will be at acceptable terms and at this time, it is not possible
to predict the outcome of these matters.
We
have incurred net losses of $18.8 million and $29.7 million for the years ended September 30, 2023 and 2022, respectively. We expect
to continue to incur significant operational expenses and net losses in the upcoming 12 months and beyond. Our net losses may fluctuate
significantly from quarter to quarter and year to year, depending on the stage and complexity of our R&D studies and related expenditures,
the receipt of additional payments on the licensing of our technology, if any, and the receipt of payments under any current or future
collaborations we may enter into.
We
have evaluated whether there are conditions or events, considered in the aggregate, that raise substantial doubt about our ability to
continue as a going concern. We believe our cash at September 30, 2023, together with the $4.1 million in net proceeds received from
our public offering of common stock and warrants on October 24, 2023, will fund our projected operations into March 2024. We also expect to receive
a $0.8 million net cash refund from the research and development tax incentive program in Australia (see Note 2 to the consolidated financial
statements) and recently received preliminary approval of our application to sell up to $4.8 million of our New Jersey state net operating
losses through the Technology Business Tax Certificate Transfer Program, subject to execution of such sale (see Note 10 to the consolidated
financial statements). Substantial
additional financing will be needed by us to fund our operations. These factors raise substantial doubt about our ability to continue
as a going concern.
The
following table summarizes our sources and uses of cash for each of the periods presented:
| | |
Year ended September 30, | |
| | |
2023 | | |
2022 | |
| Net cash used in operating activities | |
$ | (21,341,842 | ) | |
$ | (27,773,528 | ) |
| Net cash used in investing activities | |
| (443,250 | ) | |
| (810,227 | ) |
| Net cash provided by financing activities | |
| 21,006,472 | | |
| 4,014,567 | |
| Net decrease in cash | |
$ | (778,620 | ) | |
$ | (24,569,188 | ) |
Operating
Activities
During
the year ended September 30, 2023, we used $21.3 million of cash in operating activities which was primarily attributable to our net
loss of $18.8 million, a $0.5 million net increase in prepaid expenses and other assets primarily due to cash outflows for research and
development activities and a $2.4 million net decrease in accounts payable and accrued expenses primarily due to the decrease in research
and development expenses, offset by $0.3 million in acquired in-process research and development and $0.2 million in share-based compensation
expense.
During
the year ended September 30, 2022, we used $27.8 million of cash in operating activities which was primarily attributable to our net
loss of $29.7 million. This amount was offset by $0.9 million of share-based compensation expense and $1.0 million of acquired in-process
research and development, and a $0.1 million net decrease in operating assets and liabilities.
Investing
Activities
During
the year ended September 30, 2023, we used $0.4 million of cash in investing activities for the purchase of acquired in-process research
and development.
During
the year ended September 30, 2022, we used $0.8 million of cash in investing activities for the purchase of acquired in-process research
and development.
Financing
Activities
During
the year ended September 30, 2023, net cash provided by financing activities was $21.0 million, consisting primarily of net proceeds
from the sale of common stock under an at-the-market facility and through and underwritten public offering and a registered direct offering.
During
the year ended September 30, 2022, net cash provided by financing activities was $4.0 million, consisting primarily of net proceeds from
the sale of preferred stock and warrants and net proceeds from the sale of common stock.
Funding
Requirements
We
expect to continue to incur significant expenses in connection with our ongoing activities, particularly as we advance preclinical activities
and clinical trials of product candidates in development. In addition, we expect to continue to incur costs associated with operating
as a public company. The timing and amount of our operating expenditures will depend largely on:
| ● | the
scope, number, initiation, progress, timing, costs, design, duration, any potential delays,
and results of clinical trials and nonclinical studies for our current or future product
candidates; |
| | | |
| ● | the
clinical development plans we establish for these product candidates; |
| | | |
| ● | the
number and characteristics of product candidates and programs that we develop or may in-license; |
| ● | the
outcome, timing and cost of regulatory reviews, approvals or other actions to meet regulatory
requirements established by the FDA and comparable foreign regulatory authorities, including
the potential for the FDA or comparable foreign regulatory authorities to require that we
perform more studies for our product candidates than those that we currently expect; |
| | | |
| ● | our
ability to obtain marketing approval for product candidates; |
| | | |
| ● | the
cost of filing, prosecuting, defending and enforcing patent claims and other intellectual
property rights covering our product candidates; |
| | | |
| ● | our
ability to maintain, expand and defend the scope of our intellectual property portfolio,
including the cost of defending intellectual property disputes, including patent infringement
actions brought by third parties against us or our product candidates; |
| | | |
| ● | the
cost and timing of completion of commercial-scale outsourced manufacturing activities with
respect to product candidates; |
| | | |
| ● | our
ability to establish and maintain licensing, collaboration or similar arrangements on favorable
terms and whether and to what extent we retain development or commercialization responsibilities
under any new licensing, collaboration or similar arrangement; |
| | | |
| ● | the
cost of establishing sales, marketing and distribution capabilities for any product candidates
for which we may receive regulatory approval in regions where we choose to commercialize
our products on our own; |
| | | |
| ● | the
success of any other business, product or technology that we acquire or in which we invest; |
| | | |
| ● | the
costs of acquiring, licensing or investing in businesses, product candidates and technologies; |
| | | |
| ● | our
need and ability to hire additional management and scientific and medical personnel; |
| | | |
| ● | the
costs to operate as a public company in the United States, including the need to implement
additional financial and reporting systems and other internal systems and infrastructure
for our business; |
| | | |
| ● | market
acceptance of our product candidates, to the extent any are approved for commercial sale; |
| | | |
| ● | the
effect of competing technological and market developments; and |
| | | |
| ● | the
potential impact of the COVID-19 pandemic on our clinical trials and operations. |
Until
such time, if ever, as we can generate substantial product revenue, we expect to finance our cash needs through a combination of equity
offerings, debt financings, collaborations, strategic alliances, and marketing, distribution or licensing arrangements with third parties.
To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interest of ours
may be materially diluted, and the terms of such securities could include liquidation or other preferences that adversely affect the
rights of our stockholders. Debt financing and preferred equity financing, if available, may involve agreements that include restrictive
covenants that limit our ability to take specified actions, such as incurring additional debt, making capital expenditures or declaring
dividends. If we raise funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with third
parties, we may have to relinquish valuable rights to technologies, future revenue streams, research programs or product candidates or
grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings
or other arrangements when needed, we may be required to delay, reduce or eliminate product development or future commercialization efforts,
sell off assets, or grant rights to develop and market product candidates that we would otherwise prefer to develop and market.
February
2023 Offering
On
February 10, 2023, we closed a public offering of common stock and certain warrants through Chardan Capital Markets, LLC and EF Hutton,
division of Benchmark Investments LLC as underwriters, for net proceeds of $13.6 million through the issuance and sale of 530,222 shares
of our common stock and, to certain investors, pre-funded warrants to purchase 101,090 shares of common stock, and accompanying common
warrants to purchase up to an aggregate of 1,262,618 shares of our common stock (the “February Offering”). Each share of
common stock and pre-funded warrant to purchase one share of common stock was sold together with a common warrant to purchase two shares
of common stock. The public offering price of each share of common stock and accompanying common warrant was $23.76 and the public offering
price of each pre-funded warrant and accompanying common warrant was $23.7578. The common warrants are immediately exercisable at a price
of $23.76 per share of common stock, expire five years from the date of issuance and contain an alternative cashless exercise provision
whereby, subject to certain conditions, a warrant may be exercised in a cashless transaction for shares of common stock at the rate of
half a share of common stock per full share otherwise issuable upon a cash exercise. The pre-funded warrants are immediately exercisable
at any time, until exercised in full, at a price of $0.0022 per share of common stock. In addition, warrants to purchase 44,190 shares
of common stock were issued to the underwriters as compensation for their services related to the offering. These common stock warrants
have an exercise price of $29.70 per share and expire five years from the date of issuance.
June
2023 Offering
On
June 30, 2023, we closed a registered direct offering of common stock (and common stock equivalents in lieu thereof) and a concurrent
private placement of certain common stock warrants through Chardan Capital Markets, LLC as placement agent, for net proceeds of $1.9
million through the issuance and sale of 166,363 shares of our common stock and, to certain investors, pre-funded warrants to purchase
60,909 shares of common stock, and accompanying common warrants to purchase up to an aggregate of 227,272 shares of our common stock
(the “June Offering”). Each share of common stock and pre-funded warrant to purchase one share of common stock was sold together
with a common warrant to purchase one share of common stock. The public offering price of each share of common stock and accompanying
common warrant was $9.90. The common stock warrants are exercisable beginning December 30, 2023 at a price of $14.8478 per share of common
stock, expire three and half years from the date of issuance and contain an alternative cashless exercise provision. The pre-funded warrants
are immediately exercisable at any time, until exercised in full, at a price of $0.0022 per share of common stock. In addition, warrants
to purchase 6,818 shares of common stock were issued to the placement agent as compensation for its services related to the offering.
These common stock warrants have an exercise price of $14.8478 per share and expire three and a half years from the date of issuance.
October
2023 Offering
On
October 26, 2023, we closed a public offering of common stock and certain warrants through Chardan Capital Markets, LLC and Ladenburg
Thalmann & Co. Inc. as underwriters, for net proceeds of $4.1 million through the issuance and sale of 1,306,250 shares of our common
stock and, to certain investors, pre-funded warrants to purchase 1,537,500 shares of common stock, and accompanying common warrants to
purchase up to an aggregate of 5,687,500 shares of our common stock (the “October Offering”). Each share of common stock
and pre-funded warrant to purchase one share of common stock was sold together with a common warrant to purchase two shares of common
stock. The public offering price of each share of common stock and accompanying common warrant was $1.60 and the public offering price
of each pre-funded warrant and accompanying common warrant was $1.5999. The common warrants are immediately exercisable at a price of
$1.60 per share of common stock, expire five years from the date of issuance and contain an alternative cashless exercise provision.
The pre-funded warrants are immediately exercisable at any time, until exercised in full, at a price of $0.0001 per share of common stock.
In addition, warrants to purchase 85,312 shares of common stock were issued to the underwriters as compensation for their services related
to the offering. These common stock warrants have an exercise price of $2.00 per share and expire five years from the date of issuance.
At-the-Market
Offering of Common Stock
We
entered into an At-the-Market Sales Agreement with BTIG, LLC (“BTIG”) on August 15, 2022 (the “2022 Sales Agreement”).
Pursuant to the 2022 Sales Agreement, we may offer and sell, from time to time, through BTIG, as sales agent and/or principal, shares
of our common stock having an aggregate offering price of up to $25.0 million, subject to certain limitations on the amount of common
stock that may be offered and sold by us set forth in the 2022 Sales Agreement. Due to the offering limitations applicable to us, we
filed prospectus supplements for the sale of shares of our common stock for an aggregate offering price of up to $7.8 million pursuant
to the 2022 Sales Agreement. During the year ended September 30, 2023, we sold an aggregate of 136,701 shares of common stock pursuant
to the 2022 Sales Agreement with BTIG for gross proceeds of $5.8 million and net proceeds of $5.4 million. As of September 30, 2023,
there are no registered shares remaining to be sold under the 2022 Sales Agreement.
Contractual
Obligations and Commitments
The
following table summarizes our contractual obligations as of September 30, 2023 and the effects that such obligations are expected to
have on our liquidity and cash flows in future periods:
| | |
Less than 1 Year | | |
1 to 3 Years | | |
4 to 5 Years | | |
More than 5 Years | | |
Total | |
| Operating Lease (1) | |
$ | 93,614 | | |
$ | 143,703 | | |
$ | — | | |
$ | — | | |
$ | 237,317 | |
| Total | |
$ | 93,614 | | |
$ | 143,703 | | |
$ | — | | |
$ | — | | |
$ | 237,317 | |
(1)
Reflects obligations pursuant to our office lease in Princeton, New Jersey.
In
addition to the contracts with payment commitments that we have reflected in the table above, we have entered into other contracts in
the normal course of business with certain CROs, CMOs and other third-parties for preclinical research studies and testing, clinical
trials and manufacturing services. These contracts do not contain any minimum purchase commitments and are cancellable upon prior notice
and as a result, are not included in the table of contractual obligations and commitments above. Payments due upon cancellation consist
only of payments for services provided and expenses incurred, including non-cancellable obligations to our service providers, up to the
date of cancellation.
Critical
Accounting Policies and Estimates
Our
management’s discussion and analysis of financial condition and results of operations are based on our consolidated financial statements,
which have been prepared in accordance with U.S. GAAP. The preparation of these consolidated financial statements requires us to make
estimates and judgments that affect the reported amounts of assets, liabilities, and expenses and the disclosure of contingent assets
and liabilities in our financial statements. On an ongoing basis, we evaluate our estimates and judgments, including those related to
the accrual for research and development expenses. We base our estimates on historical experience, known trends and events, and various
other factors that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about
the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these
estimates under different assumptions or conditions.
While
our significant accounting policies are described in more detail in the notes to the consolidated financial statements included elsewhere
in this Form 10-K, we believe that the following accounting policies are those most critical to the judgments and estimates used in the
preparation of the consolidated financial statements.
Research
and Development Expenses
Research
and development expenses include all direct and indirect costs associated with the development of our biopharmaceutical products. These
expenses include personnel costs, consulting fees, and payments to third parties for research, development and manufacturing services.
These costs are charged to expense as incurred.
At
the end of each reporting period, we compare payments made to third-party service providers to the estimated progress toward completion
of the related project, based on the measure of progress as defined in the contract. Factors we consider in preparing the estimates include
costs incurred by the service provider, milestones achieved, and other criteria related to the efforts of our service providers. Such
estimates are subject to change as additional information becomes available. Depending on the timing of payment to the third-party service
providers and the progress we estimate has been made as a result of the service provided, we will record a prepaid expense or accrued
liability related to these costs. Contingent development or regulatory milestone payments are recognized upon the related resolution
of such contingencies. As of September 30, 2023, we did not make any material adjustments to our prior estimates of accrued research
and development expenses.
Recently
Issued Accounting Pronouncements
A
description of recently issued accounting pronouncements that may potentially impact our financial position and results of operations
is disclosed in Note 2 to the consolidated financial statements included elsewhere in this Form 10-K.
Item
7A. Qualitative and Quantitative Disclosures about Market Risk.
Not
applicable.
Item
8. Financial Statements and Supplementary Data.
SONNET
BIOTHERAPEUTICS HOLDINGS, INC.
INDEX
TO THE CONSOLIDATED FINANCIAL STATEMENTS
Report
of Independent Registered Public Accounting Firm
To the Stockholders and Board of Directors
Sonnet BioTherapeutics Holdings, Inc.:
Opinion
on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets
of Sonnet BioTherapeutics Holdings, Inc. and subsidiaries (the Company) as of September 30, 2023 and 2022, the related
consolidated statements of operations, changes in stockholders’ deficit, and cash flows for the years then ended, and the related
notes (collectively, the consolidated financial statements). In our opinion, the consolidated financial statements present fairly,
in all material respects, the financial position of the Company as of September 30, 2023 and 2022, and the results of its operations
and its cash flows for the years then ended, in conformity with U.S. generally accepted accounting principles.
Going
Concern
The accompanying consolidated financial statements have been
prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements,
the Company has incurred recurring losses and negative cash flows from operations since inception and will require substantial additional
financing to continue to fund its research and development activities that raise substantial doubt about its ability to continue as a
going concern. Management’s plans in regard to these matters are also described in Note 1. The consolidated financial statements
do not include any adjustments that might result from the outcome of this uncertainty.
Basis
for Opinion
These consolidated financial statements are the responsibility
of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our
audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are
required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and
regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of
the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial
statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged
to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding
of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s
internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks
of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond
to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated
financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management,
as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable
basis for our opinion.
Critical
Audit Matter
The critical audit matter communicated below is a matter arising
from the current period audit of the consolidated financial statements that was communicated or required to be communicated to the audit
committee and that: (1) relates to accounts or disclosures that are material to the consolidated financial statements and (2) involved
our especially challenging, subjective, or complex judgments. The communication of a critical audit matter does not alter in any way our
opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below,
providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Prepaid
development expenses and accrued research and development expense
As discussed in Notes 2 and 3 to the consolidated financial
statements, research and development costs are expensed as incurred, which include amounts due to third parties for research, development,
and manufacturing services. At the end of each reporting period, the Company compares the payments made to third-party service providers
to the estimated progress towards completion of the related project, based on the measure of progress as defined in the contract. Factors
the Company considers in preparing the estimates include costs incurred by the service provider, milestones achieved, and other criteria
related to the efforts of its service providers. Depending on the timing of payments to the third-party service providers and the progress
the Company estimates has been made as a result of the services provided, the Company will record a prepaid expense or accrued liability
related to these costs. As of September 30, 2023, the Company reported prepaid expenses and other current assets of $1.7 million, a portion
of which related to these costs, and accrued research and development expenses of $0.9 million.
We identified the evaluation of certain prepaid and accrued
research and development expenses for third- party service providers as a critical audit matter. Evaluating the estimated progress toward
completion of research and development projects, including the factors described above, required especially subjective auditor judgment.
The following are the primary procedures we performed
to address this critical audit matter. To evaluate the Company’s estimate of costs incurred as of September 30, 2023, for a selection
of prepaid and accrued research and development expenses, we (1) examined the provisions in the contracts, invoices and communications
received from third party service providers related to the project status; (2) sent confirmations to the third-party service providers;
and (3) inquired of the individuals who are responsible for monitoring and tracking the status of research and development activities
/s/ KPMG LLP
We have served as the Company’s auditor since 2015.
Philadelphia, Pennsylvania
December 14, 2023
Sonnet
BioTherapeutics Holdings, Inc.
Consolidated
Balance Sheets
| | |
2023 | | |
2022 | |
| | |
September 30, | |
| | |
2023 | | |
2022 | |
| Assets | |
| | | |
| | |
| Current assets: | |
| | | |
| | |
| Cash and cash equivalents | |
$ | 2,274,259 | | |
$ | 3,052,879 | |
| Prepaid expenses and other current assets | |
| 1,677,396 | | |
| 1,643,743 | |
| Incentive tax receivable | |
| 786,574 | | |
| 717,305 | |
| Total current assets | |
| 4,738,229 | | |
| 5,413,927 | |
| Property and equipment, net | |
| 33,366 | | |
| 46,211 | |
| Operating lease right-of-use asset | |
| 193,689 | | |
| 256,594 | |
| Deferred offering costs | |
| 49,988 | | |
| 113,280 | |
| Other assets | |
| 414,206 | | |
| — | |
| Total assets | |
$ | 5,429,478 | | |
$ | 5,830,012 | |
| Liabilities and stockholders’ deficit | |
| | | |
| | |
| Current liabilities: | |
| | | |
| | |
| Related party notes | |
$ | — | | |
$ | 748 | |
| Accounts payable | |
| 2,201,999 | | |
| 4,752,340 | |
| Accrued expenses and other current liabilities | |
| 3,230,922 | | |
| 3,193,972 | |
| Current portion of operating lease liability | |
| 73,048 | | |
| 51,328 | |
| Deferred income | |
| 18,626 | | |
| 166,431 | |
| Total current liabilities | |
| 5,524,595 | | |
| 8,164,819 | |
| Operating lease liability, net of current portion | |
| 130,863 | | |
| 203,912 | |
| Total liabilities | |
| 5,655,458 | | |
| 8,368,731 | |
| Commitments and contingencies (Note 4) | |
| - | | |
| | |
| Stockholders’ deficit: | |
| | | |
| | |
| Common stock, $0.0001 par value: 125,000,000 shares authorized; 1,750,426 and 251,955 issued and outstanding at September 30, 2023 and 2022, respectively | |
| 175 | | |
| 25 | |
| Additional paid-in capital | |
| 110,017,598 | | |
| 88,872,315 | |
| Accumulated deficit | |
| (110,243,753 | ) | |
| (91,411,059 | ) |
| Total stockholders’ deficit | |
| (225,980 | ) | |
| (2,538,719 | ) |
| Total liabilities and stockholders’ deficit | |
$ | 5,429,478 | | |
$ | 5,830,012 | |
See
accompanying notes to consolidated financial statements
Sonnet
BioTherapeutics Holdings, Inc.
Consolidated
Statements of Operations
| | |
2023 | | |
2022 | |
| | |
Years ended September 30, | |
| | |
2023 | | |
2022 | |
| Collaboration revenue | |
$ | 147,805 | | |
$ | 349,943 | |
| Operating expenses: | |
| | | |
| | |
| Research and development | |
| 11,814,690 | | |
| 21,444,019 | |
| General and administrative | |
| 7,125,732 | | |
| 8,575,283 | |
| Total operating expense | |
| 18,940,422 | | |
| 30,019,302 | |
| Loss from operations | |
| (18,792,617 | ) | |
| (29,669,359 | ) |
| | |
| | | |
| | |
| Foreign exchange loss | |
| (40,077 | ) | |
| (52,482 | ) |
| Net loss | |
$ | (18,832,694 | ) | |
$ | (29,721,841 | ) |
| Per share information: | |
| | | |
| | |
| Net loss per share, basic and diluted | |
$ | (18.14 | ) | |
$ | (150.52 | ) |
| Weighted average shares outstanding, basic and diluted | |
$ | 1,038,188 | | |
$ | 197,462 | |
See
accompanying notes to consolidated financial statements
Sonnet
BioTherapeutics Holdings, Inc.
Consolidated
Statements of Changes in Stockholders’ Equity (Deficit)
| | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
capital | | |
deficit | | |
Total | |
| | |
Preferred stock | | |
Common stock | | |
Additional paid-in | | |
Accumulated | | |
| |
| | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
capital | | |
deficit | | |
Total | |
| Balance at October 1, 2021 | |
| — | | |
$ | — | | |
| 195,579 | | |
$ | 19 | | |
$ | 83,949,046 | | |
$ | (61,689,218 | ) | |
$ | 22,259,847 | |
| Sale of preferred stock and common stock warrants, net of issuance costs | |
| 22,500 | | |
| 1,540,640 | | |
| — | | |
| — | | |
| 597,574 | | |
| — | | |
| 2,138,214 | |
| Conversion of preferred stock into common stock | |
| (22,500 | ) | |
| (1,540,640 | ) | |
| 25,101 | | |
| 3 | | |
| 1,540,637 | | |
| — | | |
| — | |
| Sale of common stock, net of issuance costs | |
| — | | |
| — | | |
| 30,206 | | |
| 3 | | |
| 1,908,690 | | |
| — | | |
| 1,908,693 | |
| Issuance of common stock on vesting of restricted stock units | |
| — | | |
| — | | |
| 1,087 | | |
| — | | |
| — | | |
| — | | |
| — | |
| Share-based compensation | |
| — | | |
| — | | |
| — | | |
| — | | |
| 876,368 | | |
| — | | |
| 876,368 | |
| Net loss | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| (29,721,841 | ) | |
| (29,721,841 | ) |
| Balance at September 30, 2022 | |
| — | | |
| — | | |
| 251,973 | | |
| 25 | | |
| 88,872,315 | | |
| (91,411,059 | ) | |
| (2,538,719 | ) |
| Balance | |
| — | | |
| — | | |
| 251,973 | | |
| 25 | | |
| 88,872,315 | | |
| (91,411,059 | ) | |
| (2,538,719 | ) |
| Sale of common stock, net of issuance costs | |
| — | | |
| — | | |
| 833,287 | | |
| 83 | | |
| 20,895,875 | | |
| — | | |
| 20,895,958 | |
| Net share settlement of warrants | |
| — | | |
| — | | |
| 519,492 | | |
| 52 | | |
| (52 | ) | |
| — | | |
| — | |
| Issuance of common stock on vesting of restricted stock units | |
| — | | |
| — | | |
| 7,676 | | |
| 1 | | |
| (1 | ) | |
| — | | |
| — | |
| Exercise of warrants | |
| — | | |
| — | | |
| 137,998 | | |
| 14 | | |
| 835 | | |
| — | | |
| 849 | |
| Share-based compensation | |
| — | | |
| — | | |
| — | | |
| — | | |
| 248,626 | | |
| — | | |
| 248,626 | |
| Net loss | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| (18,832,694 | ) | |
| (18,832,694 | ) |
| Balance at September 30, 2023 | |
| — | | |
$ | — | | |
| 1,750,426 | | |
$ | 175 | | |
$ | 110,017,598 | | |
$ | (110,243,753 | ) | |
$ | (225,980 | ) |
| Balance | |
| — | | |
$ | — | | |
| 1,750,426 | | |
$ | 175 | | |
$ | 110,017,598 | | |
$ | (110,243,753 | ) | |
$ | (225,980 | ) |
See
accompanying notes to consolidated financial statements
Sonnet
BioTherapeutics Holdings, Inc.
Consolidated
Statements of Cash Flows
| | |
2023 | | |
2022 | |
| | |
Years ended September 30, | |
| | |
2023 | | |
2022 | |
| Cash flows from operating activities: | |
| | | |
| | |
| Net loss | |
$ | (18,832,694 | ) | |
$ | (29,721,841 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | |
| | | |
| | |
| Acquired in-process research and development | |
| 282,000 | | |
| 971,477 | |
| Depreciation | |
| 12,845 | | |
| 12,845 | |
| Amortization of operating lease right-of-use asset | |
| 62,905 | | |
| 80,412 | |
| Share-based compensation | |
| 248,626 | | |
| 876,368 | |
| Changes in operating assets and liabilities: | |
| | | |
| | |
| Prepaid expenses and other current assets | |
| (33,653 | ) | |
| (454,269 | ) |
| Income tax receivable | |
| (69,269 | ) | |
| (717,305 | ) |
| Other assets | |
| (414,206 | ) | |
| — | |
| Accounts payable | |
| (2,631,215 | ) | |
| 971,041 | |
| Accrued expenses and other current liabilities | |
| 231,953 | | |
| 641,372 | |
| Operating lease liability | |
| (51,329 | ) | |
| (83,685 | ) |
| Deferred income | |
| (147,805 | ) | |
| (349,943 | ) |
| Net cash used in operating activities | |
| (21,341,842 | ) | |
| (27,773,528 | ) |
| Cash flows from investing activities: | |
| | | |
| | |
| Purchases of in-process research and development | |
| (443,250 | ) | |
| (810,227 | ) |
| Net cash used in investing activities | |
| (443,250 | ) | |
| (810,227 | ) |
| Cash flows from financing activities: | |
| | | |
| | |
| Proceeds from sale of preferred stock and common stock warrants, net of issuance costs | |
| — | | |
| 2,172,649 | |
| Proceeds from sale of common stock, net of issuance costs | |
| 21,006,371 | | |
| 1,841,918 | |
| Proceeds from exercise of warrants, net of issuance costs | |
| 849 | | |
| — | |
| Repayments of related party notes | |
| (748 | ) | |
| — | |
| Net cash provided by financing activities | |
| 21,006,472 | | |
| 4,014,567 | |
| | |
| | | |
| | |
| Net decrease in cash | |
| (778,620 | ) | |
| (24,569,188 | ) |
| Cash, beginning of year | |
| 3,052,879 | | |
| 27,622,067 | |
| Cash, end of year | |
$ | 2,274,259 | | |
$ | 3,052,879 | |
| | |
| | | |
| | |
| Supplemental disclosure of non-cash operating, investing and financing activities: | |
| | | |
| | |
| Change in operating lease right-of-use asset and liability due to amended lease | |
$ | — | | |
$ | 213,793 | |
| Deferred offering costs charged against proceeds from sale of common stock | |
$ | 32,340 | | |
$ | — | |
| Deferred offering costs in accounts payable and accrued expenses | |
$ | 49,988 | | |
$ | 80,940 | |
| Net settlement of warrants | |
$ | 52 | | |
$ | — | |
| In-process research and development in accrued expenses | |
$ | — | | |
$ | 161,250 | |
| Common stock issuance costs in accounts payable and accrued expenses | |
$ | 78,073 | | |
$ | — | |
See
accompanying notes to consolidated financial statements
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
1.
Organization and Description of Business
Description
of business
Sonnet
BioTherapeutics, Inc. (“Prior Sonnet”) was incorporated as a New Jersey corporation on April 6, 2015. Prior Sonnet completed
a merger with publicly-held Chanticleer Holdings, Inc. (“Chanticleer”) on April 1, 2020. After the merger, Chanticleer changed
its name to Sonnet BioTherapeutics Holdings, Inc. (“Sonnet” or the “Company”). Sonnet is a clinical stage, oncology-focused
biotechnology company with a proprietary platform for innovating biologic medicines of single or bifunctional action. Known as FHAB™
(Fully Human Albumin Binding), the technology utilizes a fully human single chain antibody fragment (scFv) that binds to and “hitch-hikes”
on human serum albumin (“HSA”) for transport to target tissues.
Sonnet’s
lead proprietary asset, SON-1010, is a fully human version of Interleukin 12 (“IL-12”), covalently linked to the FHAB
construct, for which Sonnet is pursuing clinical development in solid tumor indications, including ovarian cancer, non-small cell
lung cancer and head and neck cancer. In March 2022, the FDA cleared Sonnet’s Investigational New Drug (“IND”) application
for SON-1010. This allowed the Company to initiate a U.S. clinical trial (SB101) in oncology patients with solid tumors during the second
calendar quarter of 2022. In September 2021, the Company created a wholly-owned Australian subsidiary, SonnetBio Pty Ltd (“Subsidiary”),
for the purpose of conducting certain clinical trials. Sonnet received approval and initiated an Australian clinical study (SB102) of
SON-1010 in healthy volunteers during the third calendar quarter of 2022. Interim safety and tolerability data from the SB101 and SB102
studies were reported in April 2023.
In
January 2023, Sonnet announced a collaboration agreement with Roche for the clinical evaluation of SON-1010 with atezolizumab (Tecentriq®).
The companies have entered into a Master Clinical Trial and Supply Agreement (“MCSA”), along with ancillary Quality and Safety
Agreements, to study the safety and efficacy of the combination of SON-1010 and atezolizumab in a platinum-resistant ovarian cancer (“PROC”)
patient setting. Further, the companies will provide SON-1010 and atezolizumab, respectively, for use in the Phase 1b/Phase 2a combination
safety, dose-escalation, and efficacy study (SB221). Part 1 of this 2-part study was approved in June 2023 by the local Human
Research Ethics Committee in Australia under CT-2023-CTN-01399-1 and the Therapeutic Goods Administration has been notified. In August
2023, the FDA accepted the IND for SB221. The trial consists of a modified 3+3 dose-escalation design in Part 1 to establish the maximum
tolerated dose (MTD) of SON-1010 with a fixed dose of atezolizumab. Clinical benefit in PROC will be confirmed in an expansion group to
establish the recommended Phase 2 dose (RP2D). Part 2 of the study will then investigate SON-1010 monotherapy, its use in combination
with atezolizumab, or the standard of care (SOC) for PROC in a randomized comparison to show proof-of-concept (POC).
As part of the ongoing cost-cutting evaluations, all antiviral development with SON-1010 has been suspended.
The Company acquired the global development rights to its most advanced
compound, SON-080, a fully human version of Interleukin 6 (“IL-6”), in April 2020 through its acquisition of the outstanding
shares of Relief Therapeutics SA. Sonnet is advancing SON-080 in target indications of Chemotherapy-Induced Peripheral Neuropathy (“CIPN”)
and Diabetic Peripheral Neuropathy (“DPN”). Sonnet received approval to initiate an ex-U.S. Phase 1b/2a study with SON-080
in CIPN during the third quarter of 2022. The Data Safety Monitoring Board (“DSMB”) overseeing the study is expected to meet
during the first calendar quarter of 2024. Following the completion of the DSMB review, Sonnet anticipates announcing initial safety data
from the CIPN study. Pursuant to a license agreement the Company entered into with New Life Therapeutics Pte, Ltd. (“New Life”)
of Singapore in May 2021, Sonnet and New Life will be jointly responsible for developing SON-080 in DPN. The objective will be to analyze
the data and to consider initiating a Phase 2 study once the CIPN safety data has been evaluated.
SON-1210
(IL12-FHAB-IL15), Sonnet’s lead bi-specific construct, combines FHAB with fully human IL-12 and fully human
Interleukin 15 (“IL-15”). This compound is being developed for solid tumor indications, including colorectal cancer. In February
2023, the Company announced the successful completion of two IND-enabling toxicology studies with SON-1210 in non-human primates. Sonnet
is prepared to initiate the regulatory authorization process for SON-1210 pending the outcome of any partnering activity.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
SON-1410
(IL18-FHAB-IL12) is a bi-specific combination of Interleukin 18 (“IL-18”) and IL-12 for solid tumor cancers. Cell line development and process development are ongoing, with early experimental
drug supply suitable for formulation and analytical method development activities. After some delays in 2023, activities will continue
into 2024 with the potential to generate a drug suitable for preclinical studies and subsequent human studies.
The
Company has completed sequence confirmation for SON-3015 (anti-IL6-FHAB-anti-TGFβ). Early stage bi-specific drug has
been generated and is being stored for future use in in vivo mice studies. Sonnet has elected to place the SON-3015 development
program on hold for expense reduction purposes.
Liquidity
The
Company has incurred recurring losses and negative cash flows from operations since inception and it expects to generate losses from
operations for the foreseeable future primarily due to research and development costs for its potential product candidates. The Company
believes its cash of $2.3 million at September 30, 2023, in addition to the $4.1 million in net proceeds received from the October Offering
described below, will fund the Company’s projected operations into March 2024. The Company also expects to receive a $0.8 million net cash refund from
the research and development tax incentive program in Australia (see Note 2) and recently received preliminary approval of its application
to sell up to $4.8 million of its New Jersey state net operating losses through the Technology Business Tax Certificate Transfer Program
(the “Program”), subject to execution of such sale (see Note 10). Substantial additional financing will be needed
by the Company to fund its operations. These factors raise substantial doubt about the Company’s ability to continue as a going
concern. The accompanying consolidated financial statements have been prepared on a going concern basis, which contemplates the realization
of assets and satisfaction of liabilities in the normal course of business. The consolidated financial statements do not include any
adjustments that might result from the outcome of this uncertainty.
On
October 26, 2023, the Company closed a public offering of common stock and certain warrants through Chardan Capital Markets, LLC and
Ladenburg Thalmann & Co. Inc. as underwriters, for net proceeds of $4.1 million through the issuance and sale of 1,306,250 shares
of its common stock and, to certain investors, pre-funded warrants to purchase 1,537,500 shares of common stock, and accompanying common
warrants to purchase up to an aggregate of 5,687,500 shares of its common stock (the “October Offering”). Each share of common
stock and pre-funded warrant to purchase one share of common stock was sold together with a common warrant to purchase two shares of
common stock. The public offering price of each share of common stock and accompanying common warrant was $1.60 and the public offering
price of each pre-funded warrant and accompanying common warrant was $1.5999. The common warrants are immediately exercisable at a price
of $1.60 per share of common stock, expire five years from the date of issuance and contain an alternative cashless exercise provision.
The pre-funded warrants are immediately exercisable at any time, until exercised in full, at a price of $0.0001 per share of common stock.
In addition, warrants to purchase 85,312 shares of common stock were issued to the underwriters as compensation for their services related
to the offering. These common stock warrants have an exercise price of $2.00 per share and expire five years from the date of issuance.
The
Company plans to secure additional capital in the future through equity or debt financings, partnerships, collaborations, or other sources
to carry out the Company’s planned development activities. If additional capital is not available when required, the Company may
need to delay or curtail its operations until such funding is received. Various internal and external factors will affect whether and
when the Company’s product candidates become approved for marketing and successful commercialization. The regulatory approval and
market acceptance of the Company’s product candidates, length of time and cost of developing and commercializing these product
candidates and/or failure of them at any stage of the approval process will materially affect the Company’s financial condition
and future operations.
Operations
since inception have consisted primarily of organizing the Company, securing financing, developing technologies through research and
development and conducting preclinical studies. The Company faces risks associated with companies whose products are in development.
These risks include the need for additional financing to complete its research and development, achieving its research and development
objectives, defending its intellectual property rights, recruiting and retaining skilled personnel, and dependence on key members of
management.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
2.
Summary of Significant Accounting Policies
a.
Basis of presentation
The
accompanying consolidated financial statements have been prepared in conformity with U.S. generally accepted accounting principles (“U.S.
GAAP”). Any reference in these notes to applicable guidance is meant to refer to U.S. GAAP as found in the Accounting Standards
Codification (“ASC”) and Accounting Standards Updates (“ASU”) promulgated by the Financial Accounting Standards
Board (“FASB”).
b.
Consolidation
The
consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries. All intercompany accounts and
transactions have been eliminated in consolidation.
c.
Use of estimates
The
preparation of the consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions
that affect the amounts reported in the consolidated financial statements and accompanying notes. Significant estimates and assumptions
reflected in these consolidated financial statements include the accrual of research and development expenses. Estimates and assumptions
are periodically reviewed in-light of changes in circumstances, facts and experience. Changes in estimates are recorded in the period
in which they become known. Actual results could differ from management’s estimates.
d.
Reclassifications
Certain
amounts from the prior period have been reclassified to conform with the current period presentation.
e.
Segment information
Operating
segments are defined as components of an enterprise about which separate discrete information is available for evaluation by the chief
operating decision maker, or decision-making group, in deciding how to allocate resources and assess performance. The Company views its
operations and manages its business in one segment.
f.
Fair value of financial instruments
Management
believes that the carrying amounts of the Company’s financial instruments, including accounts payable, approximate fair value due
to the short-term nature of those instruments.
g.
Property and equipment
Property
and equipment are recorded at cost and depreciated using the straight-line method over the estimated useful lives of the assets. Expenditures
for repairs and maintenance that do not extend the estimated useful life or improve an asset are expensed as incurred. Upon retirement
or sale, the cost and related accumulated depreciation and amortization of assets disposed of are removed from the accounts, and any
resulting gain or loss is included in the consolidated statement of operations.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
h.
Impairment of long-lived assets
The
Company reviews long-lived assets, such as property and equipment, for impairment whenever events or changes in circumstances indicate
that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison
of the carrying amount of an asset to the undiscounted future cash flows expected to be generated by that asset. If the carrying amount
of an asset exceeds its estimated undiscounted future cash flows, then an impairment charge is recognized for the amount by which the
carrying value of the asset exceeds the estimated fair value of the asset. There were no impairment charges recorded during the fiscal
years ended September 30, 2023 and 2022.
i.
Deferred offering costs
Legal
and other costs incurred in relation to equity offerings are capitalized as deferred offering costs and charged against the proceeds
from equity offerings when received. If a financing is abandoned, deferred offering costs are expensed. As of September 30, 2023, the
Company had $49,988 in deferred offering costs associated with the October Offering. As of September 30, 2022, the Company had $0.1 million
in deferred offering costs associated with the 2022 Sales Agreement (see Note 7).
j.
Incentive tax receivable
Subsidiary
is eligible to participate in an Australian research and development tax incentive program. As part of this program, Subsidiary is eligible
to receive a cash refund from the Australian Taxation Office for a percentage of the research and development costs expended by Subsidiary
in Australia. The
cash refund is available to eligible companies with annual aggregate revenues of less than $20.0 million (Australian) during the reimbursable
period. The Company estimates the amount of cash
refund it expects to receive related to the Australian research and development tax incentive program and records the incentive when
it is probable (i) the Company will comply with relevant conditions of the program and (ii) the incentive will be received. As of both
September 30, 2023 and 2022, the Company’s estimate of the amount of cash refund it expects to receive for eligible spending related
to the Australian research and development tax incentive program was $0.8
million. For each of the years ended September
30, 2023 and 2022, $0.8
million for the expected net cash refund related to the tax
incentive program was included in research and development expenses. In January 2023, the Company received $1.1
million from the Australian government related
to eligible research and development expenses for the year ended September 30, 2022. In April 2023, the Company refunded the Australian
government for $0.2 million
due to new information on the Company’s clinical trial studies.
k.
Collaboration revenue
Collaboration
arrangements may contain multiple components, which may include (i) licenses; (ii) research and development activities; and (iii) the
manufacturing and supply of certain materials. Payments pursuant to these arrangements may include non-refundable payments, upfront payments,
milestone payments upon the achievement of significant regulatory and development events, sales milestones and royalties on product sales.
The amount of variable consideration is constrained until it is probable that the revenue is not at a significant risk of reversal in
a future period.
In
determining the appropriate amount of revenue to be recognized as the Company fulfills its obligations under a collaboration arrangement,
the Company performs the following steps: (i) identification of the promised goods or services in the contract; (ii) determination of
whether the promised goods or services are performance obligations, including whether they are capable of being distinct; (iii) measurement
of the transaction price, including the constraint on variable consideration; (iv) allocation of the transaction price to the performance
obligations; and (v) recognition of revenue as the Company satisfies each performance obligation.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
The
Company applies significant judgment when evaluating whether contractual obligations represent distinct performance obligations, allocating
transaction price to performance obligations within a contract, determining when performance obligations have been met, and assessing
the recognition of variable consideration. When consideration is received prior to the Company completing its performance obligation
under the terms of a contract, a contract liability is recorded as deferred income. Deferred income expected to be recognized as revenue
within the twelve months following the balance sheet date is classified as a current liability. In May 2021, the Company entered into
a License Agreement (the “New Life Agreement”) with New Life. See Note 6 for further discussion of the New Life Agreement.
l.
Research and development expense
Research
and development expenses include all direct and indirect costs associated with the development of the Company’s biopharmaceutical
products. These expenses include personnel costs, consulting fees, and payments to third parties for research, development, and manufacturing
services. These costs are charged to expense as incurred.
At
the end of the reporting period, the Company compares payments made to third-party service providers to the estimated progress toward
completion of the related project, based on the measure of progress as defined in the contract. Factors the Company considers in preparing
the estimates include costs incurred by the service provider, milestones achieved, and other criteria related to the efforts of its service
providers. Such estimates are subject to change as additional information becomes available. Depending on the timing of payment to the
service providers and the progress that the Company estimates has been made as a result of the service provided, the Company will record
a prepaid expense or accrued liability relating to these costs. Upfront milestone payments made to third parties who perform research
and development services on the Company’s behalf are expensed as services are rendered. Contingent development or regulatory milestone
payments are recognized upon the related resolution of such contingencies.
m.
Foreign currency
Transaction
gains and losses resulting from exchange rate changes on transactions denominated in currencies other than the U.S. dollar are included
in operations in the period in which the transaction occurs and reported within the foreign exchange loss line item in the consolidated
statements of operations.
n.
Share-based compensation
The
Company measures equity classified share-based awards granted to employees and nonemployees based on the estimated fair value on the
date of grant and recognizes compensation expense of those awards over the requisite service period, which is the vesting period of the
respective award. The Company accounts for forfeitures as they occur. For share-based awards with service-based vesting conditions, the
Company recognizes compensation expense on a straight-line basis over the service period. The Company classifies share-based compensation
expense in its consolidated statements of operations in the same manner in which the award recipient’s payroll costs are classified
or in which the award recipient’s service payments are classified.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
o.
Income taxes
The
Company uses the asset-and-liability method of accounting for income taxes. Deferred tax assets and liabilities are recognized for the
estimated future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and
liabilities and their respective tax bases, and operating loss and credit carryforwards. Deferred tax assets and liabilities are measured
using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered
or settled. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not
be realized. The Company recognizes the benefit of an uncertain tax position that it has taken or expects to take on its income tax return
if such a position is more likely than not to be sustained.
p.
Reverse stock split
On
August 31, 2023, the Company filed a Certificate of Amendment to its Certificate of Incorporation, as amended, with the Secretary of
State of the State of Delaware, which effected a 1-for
-22 reverse stock split of the Company’s issued and outstanding shares of common stock. As a result of the reverse
stock split, every 22 shares of common stock issued and outstanding was converted into one share of common stock. The reverse stock
split affected all stockholders uniformly and did not alter any stockholder’s percentage interest in the Company’s
equity. No fractional shares were issued in connection with the reverse stock split. Stockholders who would otherwise be entitled to
a fractional share of common stock were instead entitled to receive a proportional cash payment. The reverse stock split did not
change the par value or authorized number of shares of common stock. All common share and per share amounts presented in the
consolidated financial statements and accompanying notes have been retroactively adjusted to reflect the reverse stock
split.
q.
Net loss per share
Basic
net loss per share is computed by dividing net loss by the weighted-average number of shares of common stock outstanding during each
period (and potential shares of common stock that are exercisable for little or no consideration).
Diluted
loss per share includes the effect, if any, from the potential exercise or conversion of securities such as common stock warrants and
stock options which would result in the issuance of incremental shares of common stock. For diluted net loss per share, the weighted-average
number of shares of common stock is the same for basic net loss per share due to the fact that when a net loss exists, dilutive securities
are not included in the calculation as the impact is anti-dilutive.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
The
following potentially dilutive securities have been excluded from the computation of diluted shares of common stock outstanding as they
would be anti-dilutive:
Schedule of Potentially Dilutive Securities
| | |
2023 | | |
2022 | |
| | |
September
30, | |
| | |
2023 | | |
2022 | |
| Common
stock warrants August 2021 | |
| 128,500 | | |
| 128,500 | |
| Underwriter
warrants August 2021 | |
| 2,287 | | |
| 2,287 | |
| Private
warrants | |
| — | | |
| 332 | |
| Chanticleer
warrants | |
| 57 | | |
| 57 | |
| Series
C warrants | |
| 36,778 | | |
| 36,778 | |
| Series
3 warrants | |
| 12,548 | | |
| 12,548 | |
| Unvested
restricted stock units and awards | |
| 2,326 | | |
| 2,162 | |
| Common
stock warrants February 2023 | |
| 271,883 | | |
| — | |
| Underwriter
warrants February 2023 | |
| 44,190 | | |
| — | |
| Common
stock private placement warrants June 2023 | |
| 227,272 | | |
| — | |
| Placement
agent warrants June 2023 | |
| 6,818 | | |
| — | |
| Total
anti-dilutive weighted average shares | |
| 732,659 | | |
| 182,664 | |
r.
Recent accounting pronouncements
Recently
adopted
In
May 2021, the FASB issued ASU 2021-04, Earnings Per Share (Topic 260), Debt-Modifications and Extinguishments (Subtopic 470-50), Compensation-Stock
Compensation (Topic 718), and Derivatives and Hedging-Contracts in Entity’s Own Equity (Subtopic 815-40): Issuer’s Accounting
for Certain Modifications or Exchanges of Freestanding Equity-Classified Written Call Options. The amendments in ASU 2021-04 provide
guidance to clarify and reduce diversity in an issuer’s accounting for modifications or exchanges of freestanding equity-classified
written call options (for example, warrants) that remain equity classified after modification or exchange. The adoption of ASU 2021-04
on October 1, 2022 did not have any impact on the consolidated financial statements.
In
November 2021, the Financial Accounting Standards Board, or FASB, issued Accounting Standards Update No. 2021-10, Government Assistance
(Topic 832): Disclosure by Business Entities about Government Assistance, or ASU 2021-10, which improves the transparency of government
assistance received by most business entities by requiring the disclosure of: (1) the types of government assistance received; (2) the
accounting for such assistance; and (3) the effect of the assistance on a business entity’s financial statements. This guidance
is effective for financial statements issued for annual periods beginning after 15 December 2021. The adoption of the guidance on October
1, 2022 did not have a material impact on our consolidated financial statements.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
3.
Accrued Expenses and Other Current Liabilities
Accrued
expenses and other current liabilities consisted of the following:
Schedule
of Accrued Expenses and Other Current Liabilities
| | |
2023 | | |
2022 | |
| | |
September
30, | |
| | |
2023 | | |
2022 | |
| Compensation
and benefits | |
$ | 2,091,196 | | |
$ | 1,218,530 | |
| Research
and development | |
| 913,145 | | |
| 1,593,922 | |
| Professional
fees | |
| 224,031 | | |
| 378,890 | |
| Other | |
| 2,550 | | |
| 2,630 | |
| Accrued
expenses and other current liabilities | |
$ | 3,230,922 | | |
$ | 3,193,972 | |
4.
Leases
In
December 2019, the Company entered into a 36-month lease for office space in Princeton, New Jersey, which commenced February 1, 2020.
In May 2022, the Company amended the existing lease agreement in order to increase the lease term by approximately three years, which
has been accounted for as a lease modification. The operating lease right-of-use asset and liability were remeasured at the modification
date, resulting in an increase to both balances of $0.2 million.
The
components of lease expense for the years ended September 30, 2023 and 2022 are as follows:
Schedule
of Lease Expenses
| Lease
expense | |
2023 | | |
2022 | |
| Operating
lease expense | |
$ | 90,837 | | |
$ | 94,828 | |
| Variable
lease expense | |
| 5,978 | | |
| 5,339 | |
| Total
lease cost | |
$ | 96,815 | | |
$ | 100,167 | |
At
September 30, 2023, the weighted average remaining lease term was 2.6 years and the weighted average discount rate was 12%.
Cash
flow information related to operating leases for the years ended September 30, 2023 and 2022 is as follows:
Schedule of Operating Lease Liabilities
| Cash
paid for amounts included in the measurement of lease liabilities: | |
2023 | | |
2022 | |
| Operating
cash flows from operating leases | |
$ | 79,259 | | |
$ | 98,101 | |
Future
minimum lease payments under non-cancellable leases at September 30, 2023 are as follows:
Schedule of Future Minimum Lease Payments
| | |
| | |
| Fiscal
year | |
| | |
| 2024 | |
$ | 93,614 | |
| 2025 | |
| 95,487 | |
| 2026 | |
| 48,216 | |
| Total
undiscounted lease payments | |
| 237,317 | |
| Less:
imputed interest | |
| (33,406 | ) |
| Total
lease liabilities | |
$ | 203,911 | |
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
5.
Commitments and Contingencies
Legal
proceedings
From
time to time, the Company is a party to various lawsuits, claims, and other legal proceedings that arise in the ordinary course of its
business. While the outcomes of these matters are uncertain, management does not expect that the ultimate costs to resolve these matters
will have a material adverse effect on the Company’s consolidated financial position, results of operations, or cash flows.
License
agreements
In
July 2012, the Company entered into a Discovery Collaboration Agreement (the “Collaboration Agreement”) with XOMA (US) LLC
(“XOMA”), pursuant to which XOMA granted to the Company a non-exclusive, non-transferable license and/or right to use certain
materials, technologies and related information related to discovery, optimization and development of antibodies and related proteins
and to develop and commercialize products thereunder. The Company is obligated to make contingent milestone payments to XOMA totaling
$3.8 million on a product-by-product basis upon the achievement of certain development and approval milestones related to a product.
The Company has also agreed to pay XOMA low single-digit royalties on net sales of products sold by the Company. Royalties on each product
are payable on a country-by-country basis until the later of (i) a specified period of time after the first commercial sale, and (ii)
the date of expiration of the last valid claim in the last-to-expire of the issued patents covered by the Collaboration Agreement. The
first milestone was achieved in April 2022, at which time the Company incurred a $0.5 million license fee which was recorded as acquired
in-process research and development and included as research and development expenses in the consolidated statement of operations for
the year ended September 30, 2022. No license fees were incurred during the year ended September 30, 2023.
In
August 2015, the Company entered into a License Agreement (the “ARES License Agreement”) with Ares Trading, a wholly-owned
subsidiary of Merck KGaA (“ARES”). Under the terms of the ARES License Agreement, ARES has granted the Company a sublicensable,
exclusive, worldwide, royalty-bearing license on proprietary patents to research, develop, use and commercialize products using atexakin
alfa (“Atexakin”), a low dose formulation of human IL-6 in peripheral neuropathies and vascular complications. Pursuant to
the ARES License Agreement, the Company will pay ARES high single-digit royalties on net sales of products sold by the Company. Royalties
are payable on a product-by-product and country-by-country basis until the later of (i) a specified period of time after the first commercial
sale in such country, and (ii) the last date on which such product is covered by a valid claim in such country.
In
January 2019, the Company entered into a Frame Services and License Agreement (the “Cellca Agreement”) with Sartorius Stedim
Cellca GMBH (“Cellca”), pursuant to which Cellca has granted the Company a worldwide, non-exclusive, perpetual, non-transferable
license to develop, manufacture or have manufactured, use, sell, import, export and/or otherwise commercialize product based on Cellca’s
work to generate a specified transfected cell line and develop an upstream production process for such cell line. The Cellca Agreement
is effective unless terminated by either party by giving six months notice, or by giving 14 days notice if terminated for good cause.
The Company is obligated to make milestone payments to Cellca totaling up to $0.7 million upon the achievement of certain development
and approval milestones if the Buy-Out Option is not exercised. The Company has a Buy-Out Option that will be effective between the time
of completion of a clinical trial and the receipt of regulatory approval for commercialization of product. The cost to exercise the Buy-Out
Option increases on each anniversary of the commencement date of the Buy-Out Option Period, and ranges from $0.1 million to $0.6 million.
The cost to exercise the Buy-Out Option will replace the $0.6 million contingent milestone payment due upon final regulatory approval.
The first milestone was achieved in April 2022, at which time the Company incurred a $0.1 million license fees which was recorded as
acquired in-process research and development and included in research and development expenses in the consolidated statement of operations
for the year ended September 30, 2022. No license fees were incurred during the year ended September 30, 2023.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
In
October 2021, the Company entered into a Non-Exclusive License Agreement (the “Brink Agreement”) with Brink Biologics Inc.
(“Brink”), pursuant to which Brink has granted the Company a non-exclusive, non-transferable license and limited right to
sublicense certain materials and related information to develop cell-based assays for batch, quality control, stability, efficacy, potency
or any other type of assay required for production and commercialization of products. During the product development phase, the Company
was obligated to make annual product development license fee payments of approximately $0.1 million. In April 2023, the Brink Agreement
was amended, effective November 2022, to reduce the annual license fee payments to $12,000 for storage. If materials are removed from
storage during the product development phase, the annual product development license fee of approximately $0.1 million will apply. If
a product achieves commercial status, the Company is obligated to make a commercial product license fee payment of approximately $0.1
million per commercial product. The amended agreement has an initial term of one year and will automatically renew for one additional
year unless terminated or converted to a product development license. After the second year, the license will automatically convert to
a full license requiring a product development or a commercial product license fee unless the parties mutually agree to terminate the
agreement. The Company incurred $12,000 and $0.2 million in license fees during the years ended September 30, 2023 and 2022, respectively,
which were recorded as acquired in-process research and development and included in research and development expenses in the consolidated
statements of operations.
In
February 2022, the Company entered into a Biological Materials License Agreement (the “InvivoGen Agreement”) with InvivoGen
SAS (“InvivoGen”), pursuant to which InvivoGen has granted the Company a worldwide, non-exclusive license to use certain
reporter cells for research, development and/or quality control purposes. The InvivoGen Agreement has an initial term of three years
and may be extended for two additional three-year periods upon written notice by the Company and payment of an approximately €0.1
million fee per extension (approximately $0.1 million as of September 30, 2023). The Company incurred a $0.1 million license fee which
was recorded as acquired in-process research and development and included in research and development expenses in the consolidated statement
of operations for the year ended September 30, 2022. No license fees were incurred for the year ended September 30, 2023.
In
March 2022, the Company entered into a Material Transfer and License Agreement (the “ProteoNic Agreement”) with ProteoNic
B.V. (“ProteoNic”), pursuant to which ProteoNic has granted to the Company a non-exclusive, non-transferable, non-sublicensable
(except as provided for in the ProteoNic Agreement) license for certain materials, including plasmids and DNA sequences used to generate
the vectors used in the Company’s cell lines, for the Company’s use in research, development and commercialization of product.
The license will continue until terminated by either party. The Company incurred a $24,600 license fee upon obtaining the license. The
Company is obligated to make contingent milestone payments to ProteoNic totaling up to €1.2 million (approximately $1.3 million
as of September 30, 2023) upon the achievement of certain development and commercialization milestones as outlined in the ProteoNic Agreement.
The Company incurred a $24,600 license fee which was recorded as acquired in-process research and development and included in research
and development expenses in the consolidated statement of operations for the year ended September 30, 2022. No license fees were incurred
during the year ended September 30, 2023.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
Research
and development agreement
In
December 2021, the Company entered into a Research and Development Agreement (the “Navigo Agreement”) with Navigo Proteins
GmbH (“Navigo”), pursuant to which Navigo will perform specified evaluation and development procedures to evaluate certain
materials to determine their commercial potential. Under the terms of the Navigo Agreement, the Company has granted Navigo a royalty-free,
non-exclusive, worldwide, non-sublicensable, non-transferable right and license to use certain technology to perform the evaluation and
development activities, and Navigo has granted the Company (i) an exclusive, worldwide, perpetual, irrevocable, sublicensable, transferable,
royalty-free right and license to research, develop, use, sell, have sold, distribute, import or otherwise commercially exploit certain
materials, and (ii) a non-exclusive, worldwide, perpetual, sublicensable, non-transferable right and license to make or have made such
materials. The Company incurred a $0.1 million technology access fee upon execution of the Navigo Agreement, at which time it was recorded
as acquired in-process research and development and included in research and development expenses in the consolidated statement of operations
for the year ended September 30, 2022. The Company is obligated to make contingent milestone payments to Navigo, as amended in March
2023, totaling up to $1.0 million upon the achievement of certain evaluation and development milestones as outlined in the Navigo Agreement.
Certain evaluation milestones were achieved in 2023, totaling $0.3 million in license fees, which were recorded as acquired in-process
research and development and included as research and development expenses in the consolidated statement of operations for the year ended
September 30, 2023.
Employment
agreements
The
Company has entered into employment contracts with its officers and certain employees that provide for severance and continuation of
benefits in the event of termination of employment either by the Company without cause or by the employee for good reason, both as defined
in the contract. In addition, in the event of termination of employment following a change in control, as defined, either by the Company
without cause or by the employee for good reason, any unvested portion of the employee’s initial stock option grant becomes immediately
vested.
6.
Collaboration Revenue
Under
the New Life Agreement, the Company granted New Life an exclusive license (with the right to sublicense) to develop and commercialize
pharmaceutical preparations containing a specific recombinant human IL-6, SON-080 (the “Compound”) (such preparations, the
“Products”) for the prevention, treatment or palliation of diabetic peripheral neuropathy in humans (the “DPN Field”)
in Malaysia, Singapore, Indonesia, Thailand, Philippines, Vietnam, Brunei, Myanmar, Lao PDR and Cambodia (the “Exclusive Territory”).
New Life had the option to expand (1) the field of the exclusive license to include the prevention, treatment or palliation of chemotherapy-induced
peripheral neuropathy in humans (the “CIPN Field”), which option was non-exclusive and expired on December 31, 2021; and/or
(2) the territorial scope of the license to include the People’s Republic of China, Hong Kong and/or India, which option was exclusive
and expired on December 31, 2021.
The
Company will retain all rights to manufacture Compounds and Products anywhere in the world. The Company and New Life shall enter into
a follow-on supply agreement pursuant to which the Company shall supply to New Life Products for development and commercialization thereof
in the DPN Field in the Exclusive Territory on terms to be negotiated by the parties. The Company will also assist in transferring certain
preclinical and clinical development know-how that is instrumental in New Life’s ability to benefit from the license.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
New
Life will bear the cost of, and be responsible for, among other things, conducting clinical studies and additional non-clinical studies
and other developmental and regulatory activities for and commercializing Products in the DPN Field in the Exclusive Territory.
New
Life paid the Company a $0.5 million non-refundable upfront cash payment in August 2020 upon executing a letter of intent to negotiate
a license agreement and a $0.5 million non-refundable upfront cash payment in June 2021 in connection with the execution of the New Life
Agreement. New Life is also obligated to pay a non-refundable deferred license fee of an additional $1.0 million at the time of the satisfaction
of certain milestones, as well as potential additional milestone payments to the Company of up to $19.0 million subject to the achievement
of certain development and commercialization milestones. In addition, during the Royalty Term (as defined below), New Life is obligated
to pay the Company tiered double-digit royalties ranging from 12% to 30% based on annual net sales of Products in the Exclusive Territory.
The “Royalty Term” means, on a Product-by-Product and a country-by-country basis in the Exclusive Territory, the period commencing
on the date of the first commercial sale (subject to certain conditions) of such Product in such country in the Exclusive Territory and
continuing until New Life ceases commercialization of such Product in the DIPN Field.
The
New Life Agreement will remain in effect on a Product-by-Product, country-by-country basis and will expire upon the expiration of the
Royalty Term for the last-to-expire Product in the last-to-expire country, subject to (i) each party’s early termination rights
including for material breach or insolvency or bankruptcy of the other party and (ii) the Company’s Buy Back Right and New Life’s
Give Back Right (as defined below).
In
addition, New Life granted to the Company an exclusive option to buy back the rights granted by the Company to New Life and the Company
granted New Life the right to give back the rights with respect to Products in the DPN Field in one or more countries in the Exclusive
Territory on terms to be agreed upon, which options will expire upon the initiation of a Phase III Trial for the applicable Product.
Revenue
recognition
The
Company first assessed the New Life Agreement under ASC 808, Collaborative Arrangements (“ASC 808”), to determine
whether the New Life Agreement or units of accounts within the New Life Agreement represent a collaborative arrangement based on the
risks and rewards and activities of the parties. The Company applied relevant guidance from ASC 606, Revenue from Contracts with Customers
(“ASC 606”), to evaluate the appropriate accounting for the collaborative arrangement with New Life. In accordance with
this guidance, the Company identified the following obligations under the arrangement: (i) License to develop, market, import, use and
commercialize the Product in the Field in the Exclusive Territory (the “License”); and (ii) transfer of know-how and clinical
development and regulatory activities (“R&D Activities”). The options to expand the CIPN Field and territory as well
as the future supply agreement represent optional purchases, which are accounted for as separate contracts. The Company evaluated these
separate contracts and did not identify any material right to be present. The Company determined that License and the R&D services
are not distinct from each other and therefore combined these material promises into a single performance obligation.
The
Company determined the initial transaction price of the single performance obligation to be $1.0 million, as the future development and
commercialization milestones, which represent variable consideration, are subject to constraint at inception. At the end of each subsequent
reporting period, the Company will reevaluate the probability of achievement of the future development and commercialization milestones
subject to constraint and, if necessary, will adjust its estimate of the overall transaction price. Any such adjustments will be recorded
on a cumulative catch-up basis. For the sales-based royalties, the Company will recognize revenue when the related sales occur.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
Collaboration
revenue from the single performance obligation is being recognized over the estimated performance of the R&D services. The Company
recognized $0.1 million and $0.3 million of collaboration revenue for the years ended September 30, 2023 and 2022, respectively.
7.
Stockholders’ Deficit
2023
events
The
Company entered into an At-the-Market Sales Agreement with BTIG, LLC (“BTIG”) on August 15, 2022 (the “2022 Sales Agreement”).
Pursuant to the 2022 Sales Agreement, the Company could offer and sell, from time to time, through BTIG, as sales agent and/or principal,
shares of its common stock having an aggregate offering price of up to $25.0 million, subject to certain limitations on the amount of
common stock that may be offered and sold by the Company set forth in the 2022 Sales Agreement. Due to the offering limitations applicable
to the Company, the Company filed prospectus supplements for the sale of shares of its common stock for an aggregate offering price of
up to $7.8 million pursuant to the 2022 Sales Agreement. During the year ended September 30, 2023, the Company sold an aggregate of 136,702
shares of common stock pursuant to the 2022 Sales Agreement with BTIG for gross proceeds of $5.7 million and net proceeds of $5.5 million.
There are no registered shares remaining to be sold under the 2022 Sales Agreement.
On
February 10, 2023, the Company closed a public offering of common stock and certain warrants through Chardan Capital Markets, LLC and
EF Hutton, division of Benchmark Investments LLC as underwriters, for gross proceeds of $15.0 million and net proceeds of $13.6 million
through the issuance and sale of 530,222 shares of its common stock and, to certain investors, pre-funded warrants to purchase 101,090
shares of common stock, and accompanying common warrants to purchase up to an aggregate of 1,262,618 shares of its common stock (the
“February Offering”). Each share of common stock and pre-funded warrant to purchase one share of common stock was sold together
with a common warrant to purchase two shares of common stock. The public offering price of each share of common stock and accompanying
common warrant was $23.76 and the public offering price of each pre-funded warrant and accompanying common warrant was $23.7578.
The
common stock warrants are immediately exercisable at a price of $23.76 per share of common stock, expire five years from the date of
issuance and contain an alternative cashless exercise provision whereby, subject to certain conditions, a warrant may be exercised in
a cashless transaction for shares of common stock at the rate of half a share of common stock per full share otherwise issuable upon
a cash exercise. The pre-funded warrants are immediately exercisable at any time, until exercised in full, at a price of $0.0022 per
share of common stock.
In
addition, warrants to purchase 44,190 shares of common stock were issued to the underwriters as compensation for their services related
to the offering. These common stock warrants have an exercise price of $29.70 per share and expire five years from the date of issuance.
On
June 30, 2023, the Company closed a registered direct offering of common stock (and common stock equivalents in lieu thereof) and a concurrent
private placement of certain common stock warrants through Chardan Capital Markets, LLC as placement agent, for gross proceeds of $2.3
million and net proceeds of $1.9 million through the issuance and sale of 166,363 shares of its common stock and, to certain investors,
pre-funded warrants to purchase 60,909 shares of common stock, and accompanying common warrants to purchase up to an aggregate of 227,272
shares of its common stock (the “June Offering”). Each share of common stock and pre-funded warrant to purchase one share
of common stock was sold together with a common warrant to purchase one share of common stock. The public offering price of each share
of common stock and accompanying common warrant was $9.90.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
The
common stock warrants are exercisable beginning December 30, 2023 at a price of $14.8478 per share of common stock, expire three and
half years from the date of issuance and contain an alternative cashless exercise provision. The pre-funded warrants are immediately
exercisable at any time, until exercised in full, at a price of $0.0022 per share of common stock. All of the pre-funded warrants have
been exercised.
In
addition, warrants to purchase 6,818 shares of common stock were issued to the placement agent as compensation for its services related
to the offering. These common stock warrants have an exercise price of $14.8478 per share and expire three and a half years from the
date of issuance.
2022
events
In
August 2022, the Company entered into a securities purchase agreement (the “Preferred SPA”) with several accredited investors
for the issuance and sale of (i) an aggregate of 22,275 shares of its Series 3 Convertible Preferred Stock, stated value $100 per share,
(ii) 225 shares of its Series 4 Convertible Preferred Stock, stated value $100 per share, and (iii) Series 3 warrants to purchase up
to 12,548 shares of its common stock in a private placement for aggregate gross proceeds of $2.3 million, with $0.1 million of issuance
costs for net proceeds of $2.1 million. The shares of Series 3 Convertible Preferred Stock were convertible into an aggregate of 24,850
shares of common stock of the Company and the shares of Series 4 Convertible Preferred Stock were convertible into an aggregate of 251
shares of common stock of the Company, in each case, at a conversion price of $89.628 per share. The Series 3 Warrants have an exercise
price of $89.628 per share, are exercisable commencing six months after issuance, and will expire five years from the issuance date.
The net proceeds from the private placement were allocated to the Series 3 Convertible Preferred Stock, Series 4 Convertible Preferred
Stock and Series 3 warrants based on their relative fair values.
The
shares of the Series 3 and Series 4 Convertible Preferred Stock had no voting rights other than the right to vote with common stockholders,
as a single class, on a proposed amendment to the Company’s Certificate of Incorporation, as amended, to effect a reverse split
of the outstanding shares of common stock. Each share of Series 3 Convertible Preferred Stock was entitled to vote on an as-converted
basis, and each share of Series 4 Convertible Preferred Stock was entitled to 811,688 votes per share, provided that the Series 4 Convertible
Preferred Stock be automatically voted in the same proportions as the shares of common stock and Series 3 Convertible Preferred Stock
on the reverse split proposal. The shares of the Series 3 and Series 4 Convertible Preferred Stock were convertible at the option of
the holder at any time following the effective date of a reverse split of the Company’s outstanding common stock. In addition,
the Company could elect mandatory conversion on the date of the reverse stock split or the Company could force conversion of all or a
part of the Series 3 and Series 4 Convertible Preferred Stock at any time after 120 days following the issuance of the preferred stock,
in either case provided certain equity conditions were met.
The
private placement closed on August 15, 2022, and the stockholders voted to approve a 1-for-14 reverse stock split on September 15, 2022.
The Series 3 and Series 4 Convertible Preferred Stock were converted to 25,101 shares of common stock on September 30, 2022. As of September
30, 2023 and 2022, there were no shares of preferred stock issued and outstanding.
During
the year ended September 30, 2022, the Company sold an aggregate of 30,206 shares of common stock pursuant to the 2022 Sales Agreement
with BTIG for gross proceeds of $2.0 million and net proceeds of $1.9 million.
Also
during the year ended September 30, 2022, the Company issued 1,087 shares of common stock upon the vesting of restricted stock units.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
Common
stock warrants
As
of September 30, 2023, the following equity-classified warrants and related terms were outstanding:
Schedule
of Warrants Outstanding
| | |
Warrants
Outstanding | | |
Exercise
Price | | |
Expiration
Date |
| Common
stock warrants August 2021 | |
| 128,500 | | |
$ | 261.80 | | |
August
24, 2024 |
| Underwriter
warrants August 2021 | |
| 2,287 | | |
$ | 327.25 | | |
August
19, 2024 |
| Chanticleer
warrants | |
| 57 | | |
$ | 18,018.00
-$28,028.00 | | |
April
30, 2027 - December 17, 2028 |
| Series
C warrants | |
| 36,778 | | |
$ | 982.52 | | |
October
16, 2025 |
| Series
3 warrants | |
| 12,548 | | |
$ | 89.628 | | |
August
15, 2027 |
| Common
stock warrants February 2023 | |
| 271,883 | | |
$ | 23.76 | | |
February
10, 2028 |
| Underwriter
warrants February 2023 | |
| 44,190 | | |
$ | 29.70 | | |
February
8, 2028 |
| Common
stock private placement warrants June 2023 | |
| 227,272 | | |
$ | 14.8478 | | |
December
30, 2026 |
| Placement
agent warrants June 2023 | |
| 6,818 | | |
$ | 14.8478 | | |
December
30, 2026 |
| Total | |
| 730,333 | | |
| | | |
|
During
the year ended September 30, 2023, 1,014,872 warrants were net share settled, resulting in the issuance of 519,492 shares of common stock.
During
the year ended September 30, 2023, 137,999 warrants were exercised on a cash basis. The Company received de minimus proceeds in exchange
for the issuance of 137,998 shares of common stock.
During
the year ended September 30, 2023, 332 of private warrants expired.
8.
Share-Based Compensation
In
April 2020, the Company adopted the 2020 Omnibus Equity Incentive Plan (the “Plan”). On January 1, 2023, the total number
of shares authorized under the Plan increased to 14,480. There were 14,480 shares available for issuance under the Plan as of September
30, 2023. The Plan increases the amount of shares issuable under the Plan by four percent of the outstanding shares of common stock at
each January 1, each year. The Plan permits the granting of share-based awards, including stock options, restricted stock units and awards,
stock appreciation rights and other types of awards as deemed appropriate, in each case, in accordance with the terms of the Plan. The
terms of the awards are determined by the Company’s Board of Directors.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
Restricted
stock units and awards
In
July of 2020, 2,115 restricted stock units (“RSUs”) were granted, 50% of which vested on April 2, 2021 and the remaining
50% vested on April 2, 2022. In March of 2021, an additional 152 RSUs were granted, 50% of which vested on March 25, 2022 and the remaining
50% vested on March 25, 2023. In December of 2021, 2,103 RSUs were granted, 100% of which vested on January 1, 2023. In December of 2022,
7,840 RSUs were granted, 100% of which vest on January 1, 2024.
In
January 2023, 5,514 of the RSUs granted in December 2022 were cancelled and subsequently reissued as restricted shares of the Company’s
common stock (“Restricted Stock Awards” or “RSAs”). The RSAs have the same vesting conditions as the original
RSUs issued in December 2022. The Company accounted for this as a stock compensation modification resulting in $38,837 of incremental
expense which will be recognized over the remaining vesting period.
Any
unvested RSUs or RSAs will be forfeited upon termination of services. The fair value of an RSU or RSA is equal to the fair market value
of the Company’s common stock on the date of grant. RSU and RSA expense is amortized straight-line over the vesting period.
The
Company recorded share-based compensation expense associated with the RSUs and RSAs in its accompanying consolidated statements of operations
as follows:
Schedule
of Share-based Compensation Expense
| | |
2023 | | |
2022 | |
| | |
Years
ended September 30, | |
| | |
2023 | | |
2022 | |
| Research
and development | |
$ | 121,265 | | |
$ | 437,921 | |
| General
and administrative | |
| 127,361 | | |
| 438,447 | |
| Share
Based Compensation | |
$ | 248,626 | | |
$ | 876,368 | |
The
following table summarizes RSU activity under the Plan:
Schedule
of Restricted Stock Units Activity
| | |
| | |
Weighted | |
| | |
| | |
Average
Grant | |
| | |
RSU | | |
Date
Fair Value | |
| Unvested
balance at October 1, 2021 | |
| 1,179 | | |
$ | 1,068.32 | |
| Granted | |
| 2,103 | | |
$ | 155.54 | |
| Vested | |
| (1,096 | ) | |
$ | 1,091.20 | |
| Forfeited | |
| (24 | ) | |
$ | 1,118.04 | |
| Unvested
balance at October 1, 2022 | |
| 2,162 | | |
$ | 175.56 | |
| Granted | |
| 7,840 | | |
$ | 21.47 | |
| Vested | |
| (2,162 | ) | |
$ | 12.55 | |
| Forfeited | |
| (5,514 | ) | |
$ | 21.34 | |
| Unvested
balance at September 30, 2023 | |
| 2,326 | | |
$ | 21.78 | |
As
of September 30, 2023, total unrecognized compensation expense relating to unvested RSUs granted was $12,194, which is expected to be
recognized over a weighted-average period of three months.
The
following table summarizes RSA activity under the Plan:
Schedule
of Restricted Stock Awards Activity
| | |
| | |
Weighted | |
| | |
| | |
Average
Grant | |
| | |
RSA | | |
Date
Fair Value | |
| Unvested
balance at October 1, 2022 | |
| — | | |
| — | |
| Granted | |
| 5,514 | | |
$ | 28.27 | |
| Unvested
balance at September 30, 2023 | |
| 5,514 | | |
$ | 28.27 | |
As
of September 30, 2023, total unrecognized compensation expense relating to unvested RSAs granted was $37,812, which is expected to be
recognized over a weighted-average period of three months.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
9.
Income Taxes
As
of September 30, 2023, the Company had $103.6
million, $69.5
million and $8.9
million of federal, state and foreign net operating losses, respectively. The federal and state net operating losses will begin to
expire in 2030 and the foreign net operating losses begin to expire
in 2027. As of September 30, 2023, the Company has federal and state research and development tax credit carryforwards of
$2.4
million and $0.8
million available to reduce future tax liabilities which will begin to expire in 2035 and 2030, respectively. Realization of the
deferred tax asset is contingent on future taxable income and based upon the level of historical losses, management has concluded
that the deferred tax asset does not meet the more-likely-than-not threshold for realizability. Accordingly, a full valuation
allowance continues to be recorded against the Company’s deferred tax assets as of September 30, 2023 and 2022. The valuation
allowance increased $5.8
million during the year ended September 30, 2023 and $8.3
million during the year ended September 30, 2022.
Due
to the change in ownership provisions of the Internal Revenue Code, the availability of the Company’s net operating loss carryforwards
may be subject to annual limitations, against taxable income in future periods, which could substantially limit the eventual utilization
of such carryforwards. The Company has not analyzed the historical or potential impact of its equity financings on beneficial ownership
and therefore no determination has been made whether the net operating loss carryforwards are subject to any Internal Revenue Code Section
382 limitation. To the extent there is a limitation, there would be a reduction in the deferred tax assets with an offsetting reduction
in the valuation allowance.
When
uncertain tax positions exist, the Company recognizes the tax benefit of tax positions to the extent that the benefit will more likely-than-not
be realized. The determination as to whether the tax benefit will more-likely-than-not be realized is based upon the technical merits
of the tax position as well as consideration of the available facts and circumstances. The Company recognizes interest and penalties
accrued on any unrecognized tax benefits within the provision for income taxes in its consolidated statements of operations. No unrecognized
tax benefits have been recorded.
The
tax effects of the temporary differences that gave rise to deferred taxes were as follows:
Schedule of Deferred Tax Assets and Liabilities
| | |
2023 | | |
2022 | |
| | |
September
30, | |
| | |
2023 | | |
2022 | |
| Deferred
tax assets: | |
| | | |
| | |
| Net
operating loss carryforwards | |
$ | 27,996,751 | | |
$ | 25,858,311 | |
| Research
and development credit carryforwards | |
| 3,106,675 | | |
| 2,151,942 | |
| Amortization | |
| 4,692,227 | | |
| 2,897,388 | |
| Share-based
compensation | |
| 226 | | |
| 66,832 | |
| Operating
lease liability | |
| 57,319 | | |
| 71,748 | |
| Accrued
expenses and other | |
| 546,612 | | |
| 314,210 | |
| Section
163(j) disallowed interest expense | |
| 763,172 | | |
| — | |
| Property
and equipment | |
| — | | |
| 4,790 | |
| Gross
deferred tax assets | |
| 37,162,982 | | |
| 31,365,221 | |
| Less:
valuation allowance | |
| (37,100,582 | ) | |
| (31,293,092 | ) |
| Deferred tax assets | |
| 62,400 | | |
| 72,129 | |
| Deferred
tax liabilities: | |
| | | |
| | |
| Property
and equipment | |
| (7,954 | ) | |
| — | |
| Operating
lease right-of-use asset | |
| (54,446 | ) | |
| (72,129 | ) |
| Net
deferred tax assets | |
$ | — | | |
$ | — | |
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
The
Company recorded no income tax expense or benefit for the years ended September 30, 2023 and 2022. A reconciliation of income tax (expense)
benefit at the statutory federal income tax rate and income taxes as reflected in the consolidated financial statements is as follows:
Schedule of Effective Income Tax Rate Reconciliation
| | |
2023 | | |
2022 | |
| | |
Years
ended September 30, | |
| | |
2023 | | |
2022 | |
| U.S.
federal statutory rate | |
| (21.0 | )% | |
| (21.0 | )% |
| State
taxes, net of federal benefit | |
| (7.1 | ) | |
| (6.0 | ) |
| Change
in valuation allowance | |
| 30.8 | | |
| 28.1 | |
| Research
and development credit | |
| (5.1 | ) | |
| (3.0 | ) |
| Permanent
differences | |
| (1.6 | ) | |
| 0.7 | |
| Foreign
tax rate differential | |
| 0.3 | | |
| 0.8 | |
| State NOLs | |
| 3.7 | | |
| — | |
| Other | |
| 0.0 | | |
| 0.4 | |
| Effective
income tax rate | |
| — | % | |
| — | % |
In
August 2022, the U.S. enacted the Inflation Reduction Act of 2022 (“IRA”). The IRA contains a number of tax-related provisions
that will be effective for tax years beginning after December 31, 2022, including a corporate alternative minimum tax of 15% on certain
large corporations and an excise tax of 1% on corporate stock repurchases. The Company is currently evaluating the various provisions
of the IRA and does not anticipate a material impact on its consolidated financial statements.
10.
Subsequent Events
The
Company has evaluated subsequent events from the balance sheet date through December 14, 2023, the date at which the consolidated financial statements
were available to be issued. Refer to Note 1 for information regarding the October Offering.
In
December 2023, the Company received preliminary approval of its application to sell up to $4.8 million of its New Jersey state net operating
losses through the Technology Business Tax Certificate Transfer Program (the “Program”). As outlined in the Program, although
the sale has been approved and a buyer identified, the Company must still execute an arrangement with such buyer to consummate a sale
of the state net operating losses.
Item
9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure.
None.
Item
9A. Controls and Procedures.
Conclusions
Regarding the Evaluation of Our Disclosure Controls
We
maintain disclosure controls and procedures that are designed to provide reasonable assurance that material information required to be
disclosed in our periodic reports filed under the Securities Exchange Act of 1934, as amended (the “Exchange Act”) is recorded,
processed, summarized, and reported within the time periods specified in the SEC’s rules and forms and to provide reasonable assurance
that such information is accumulated and communicated to our management, our Chief Executive Officer and our Chief Financial Officer,
to allow timely decisions regarding required disclosure. We carried out an evaluation, under the supervision and with the participation
of our management, including our principal executive and principal financial officer, of the effectiveness of the design and operation
of our disclosure controls and procedures, as defined in Rule 13(a)-15(e) under the Exchange Act. Based on this evaluation, our principal
executive officer and principal financial officer concluded that, as of September 30, 2023, our disclosure controls and procedures were
effective.
Management’s
Annual Report on Internal Control Over Financial Reporting
Our
management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f)
and 15d-15(f) under the Exchange Act. Internal control over financial reporting is a process designed to provide reasonable assurance
of the reliability of financial reporting and of the preparation of financial statements for external reporting purposes, in accordance
with U.S. generally accepted accounting principles.
Internal
control over financial reporting includes policies and procedures that (1) pertain to the maintenance of records that, in reasonable
detail, accurately and fairly reflect transactions and disposition of assets; (2) provide reasonable assurance that transactions are
recorded as necessary to permit preparation of financial statements in accordance with U.S. generally accepted accounting principles,
and that receipts and expenditures are being made only in accordance with the authorization of its management and directors; and (3)
provide reasonable assurance regarding the prevention or timely detection of unauthorized acquisition, use or disposition of our assets
that could have a material effect on tis financial statements.
Under
the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted
an evaluation of the effectiveness of our internal control over financial reporting based on criteria established in the framework in
Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission.
Based on this evaluation, our management concluded that our internal control over financial reporting was effective as of September 30,
2023.
Because
of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of
any evaluation of effectiveness to future periods are subject to the risks that controls may become inadequate because of changes in
conditions, or that the degree of compliance with the policies or procedures may deteriorate.
This
Annual Report does not include an attestation report of our independent registered public accounting firm because we are a “non-accelerated
filer,” and may take advantage of certain exemptions from various reporting requirements that are applicable to public companies
that are accelerated filers, including, but not limited to, not being required to comply with the auditor attestation requirements of
Section 404(b) of the Sarbanes-Oxley Act.
Changes
in Internal Controls over Financial Reporting
There
was no change in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f)) under the Exchange Act)
that occurred during the fourth quarter ended September 30, 2023, that has materially affected, or is reasonably likely to materially
affect, our internal control over financial reporting.
Item
9B. Other Information.
On December 11, 2023, the Compensation
Committee (the “Committee”) of the Board of Directors of Sonnet BioTherapeutics Holdings, Inc. (the “Company”),
approved restricted stock unit (“RSU”) awards (using the Company’s standard form of RSU award agreement, a copy of which
is filed as Exhibit 10.10 hereto and incorporated by reference herein) to certain executive officers of the Company under the Company’s
2020 Omnibus Equity Incentive Plan (the “Plan”). Each RSU represents a right to receive one share of the Company’s common
stock upon vesting, subject to the terms and conditions described below. The grants were made effective January 1, 2024, subject to the
availability of shares for grant under the Plan, and contingent upon the respective grantee being continuously employed by the Company
on such date; provided that each grantee may notify the Chief Executive Officer of the Company on or before December 31, 2023 if such
grantee elects to receive a restricted stock award instead of RSUs, in which case such grantee will receive an award of restricted stock
on the Company’s previously approved form.
Pankaj Mohan, Ph.D., Chief Executive Officer, received an RSU award representing 60,433 shares of the Company’s
common stock. Jay Cross, Chief Financial Officer, received an RSU award representing 11,622 shares of the Company’s common stock.
John Cini, Chief Scientific Officer received an RSU award representing 15,108 shares of the Company’s common stock. Susan Dexter,
Chief Technology Officer received an RSU award representing 11,622 shares of the Company’s common stock. Rick Kenney, Chief Medical
Officer received an RSU award representing 10,169 shares of the Company’s common stock. Each RSU award will vest as to 100% on January
1, 2025. In the event that a grantee ceases to be in Continuous Service (as defined in the Plan), any RSUs that have not vested as of
the date of cessation of service shall be forfeited.
Item
9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.
Not
applicable.
PART
III
Item
10. Directors, Executive Officers and Corporate Governance.
Directors
The
following table sets forth certain information about the current directors of the Company. Directors are elected to hold office until
the next annual meeting of stockholders and until their successors are elected and qualified.
| Directors |
|
Age |
|
Year
First
Became
Director |
| Pankaj
Mohan, Ph.D |
|
59 |
|
2020 |
| Nailesh
Bhatt |
|
51 |
|
2020 |
| Albert
Dyrness |
|
61 |
|
2020 |
| Donald
Griffith |
|
75 |
|
2020 |
| Raghu
Rao |
|
61 |
|
2020 |
| Lori
McNeill |
|
51 |
|
2022 |
Set
forth below are brief biographical descriptions of the individuals currently serving as the Company’s directors, based on information
furnished to the Company by such individuals.
Pankaj
Mohan, Ph.D.
Pankaj
Mohan, Ph.D. founded Sonnet in 2015 and has since served as a member of its board of directors, and was appointed to our Board of Directors
(the “Board”) as Chairman at the closing of the Merger. Dr. Mohan became the Chairman of Sonnet in June 2018 and the Chief
Executive Officer of Sonnet in January 2019 and was appointed President and Chief Executive Officer of the Company at the closing of
the Merger. From January 2011 to June 2018, he served as the President, Chief Executive Officer and Chairman of Oncobiologics, Inc. (now
Outlook Therapeutics, Inc.; Nasdaq: OTLK), a company that he founded in 2011. Previously, Dr. Mohan served as head of Business Operations
and Portfolio Management of Biologics Process and Product Development at Bristol-Myers Squibb Company and as a Director of Bioprocess
Engineering at Genentech, Inc. Prior to that, Dr. Mohan served as a senior manager at Eli Lilly and Company. From May 1993 to April 1996,
Dr. Mohan served as Assistant Professor (Lecturer/Fellow) at the Advanced Centre for Biochemical Engineering, University College London,
London, United Kingdom. Dr. Mohan received a Ph.D. in Biochemical Engineering from the School of Chemical Engineering, University of
Birmingham, Birmingham, United Kingdom, a Masters in Financial Management from Middlesex University Business School, London, United Kingdom,
an Executive Management Program (AMP) from Fuqua School of Business at Duke University and a Bachelor of Chemical Engineering from the
Indian Institute of Technology in Roorkee, India. He is also an author of an industry reference book on bioprocess operations (McGraw
Hill). The Company believes Dr. Mohan is capable of making valuable contributions to the Board due to his extensive knowledge of the
biopharmaceutical industry and his prior experience as an executive officer.
Nailesh
Bhatt
Nailesh
Bhatt has served on Sonnet’s board of directors since July 2018, and was appointed to our Board at the closing of the Merger. Since
January 2018, Mr. Bhatt has been the Chief Executive Officer and a Board Member of VGYAAN Pharmaceuticals LLC, a company focused on developing
and commercializing clinically critical drugs. Prior to that, in November 2001, Mr. Bhatt founded Proximare and is its Managing Director.
Proximare is a strategic advisory firm focused exclusively on the pharmaceutical industry. Mr. Bhatt also serves as Board Member of Azurity
Pharmaceuticals, Inc. since April 2018. In June 2015, Mr. Bhatt founded Proximare Lifesciences Fund. Mr. Bhatt pursued a Bachelor of
Arts at Boston University with a major in Biology. The Company believes Mr. Bhatt can make valuable contributions to the Board due to
his years of experience in the pharmaceutical industry working with start-ups to Fortune 500 companies.
Albert
Dyrness
Albert
Dyrness has served on Sonnet’s board of directors since October 2019, and was appointed to our Board at the closing of the Merger.
Mr. Dyrness is a recognized biopharmaceutical industry expert in bio-process engineering with expertise in upstream, downstream, and
fill/finish processes. Since July 2019, Mr. Dyrness has been the Managing Director of ADVENT Engineering Services, Inc., a Trinity Consultants
Company, which serves as its life-sciences division. In 1988, Mr. Dyrness Co-Founded ADVENT Engineering Services, Inc., an engineering
consulting firm serving the energy and life sciences industries. Starting with only 4 employees in the San Francisco Bay Area, ADVENT
has grown to a staff of over 130 engineers with offices in Toronto, Canada, Singapore, Raleigh, North Carolina, Portland Oregon, Boston,
Massachusetts, Irvine and San Ramon, California. In 2016, Mr. Dyrness became President and Chief Technical Officer of ADVENT and, in
2017, guided the company to a merger with Trinity Consultants, a 700-person engineering consulting firm. He also served as a member of
the board of directors of Oncobiologics, Inc. (now Outlook Therapeutics, Inc.; Nasdaq: OTLK) from December 2015 to September 2017. In
1986, Mr. Dyrness graduated from the Massachusetts Institute of Technology where he studied mechanical engineering and entrepreneurism.
The Company believes Mr. Dyrness is capable of making valuable contributions to the Board due to his years of experience in a Nasdaq-listed
public company, along with years of entrepreneurial experience, including in the biopharmaceutical industry.
Donald
Griffith, CPA
Donald
Griffith, CPA has served on Sonnet’s board of directors since its inception in April 2015, was Chairman of the Sonnet board from
April 2015 to June 2018, and was appointed to our Board at the closing of the Merger. Mr. Griffith has served as Sonnet’s Financial
Controller since January 1, 2019, and since the Merger serves as our Controller. Prior to being Financial Controller, he served as Sonnet’s
Chief Executive Officer and Chief Financial Officer from April 2015 to December 2016. Before that, Mr. Griffith was the Chief Financial
Officer, Director and Secretary of Oncobiologics Inc. (now Outlook Therapeutics; Nasdaq: OTLK) from 2011 to 2018. Mr. Griffith has over
40 years’ experience in finance and accounting and is the founder and Partner of Stolz & Griffith, LLC, a New Jersey accounting
firm. The Company believes Mr. Griffith is capable of making valuable contributions to the Board due to his years of experience in finance
as well as in the pharmaceutical industry.
Raghu
Rao
Raghu
Rao has served on Sonnet’s board of directors since November 2019, and was appointed to our Board at the closing of the Merger.
Mr. Rao is a serial entrepreneur, strategic business advisor and angel investor. Mr. Rao has founded, scaled and had successful exits
with several high-technology companies. In his 33-year career, Mr. Rao has advised clients on the strategy and roll-out of high-profile
projects, such as USA.gov, TSA Screening Gateway, Cancer.gov and other eGovernment initiatives. As the Vistage Princeton Chair, from
July 2012 to March 2017, Mr. Rao ran three high-performing peer advisory boards for middle-market CEOs and business leaders of companies
with total revenues exceeding $2 Billion. As the Chairman & President of InfoZen from August 1995 to July 2008, Mr. Rao has managed
over $1 Billion in U.S. Federal Government contracts. Mr. Rao is a 20-year Charter Member of The Indus Entrepreneurs (TiE.org) and a
5-year patron of the Indiaspora. He has held board positions at several companies including Cellix BioSciences (Jan 2016 - Jan 2017),
Paper Battery Company (Jan 2009 - Dec 2018), Kovid Group (Feb 2016 - Oct 2017) , WizNucleus (Jun 2010 - present) and InfoZen (Aug 1995
- Jul 2008). Mr. Rao is active in social entrepreneurship and community service. He co-founded the Hindu Jewish Coalition in December
2012 and Forum for Religious Freedom in March 2007, to preserve religious diversity worldwide. He has held non-profit board positions
at the Infinity Foundation (New Jersey), Arsha Vidya Gurukulam (Pennsylvania) and the Family Services Agency (Maryland). Mr. Rao has
an MBA in Finance from George Washington University (Dec 1991), an M.S. in Computer Science from Virginia Tech (Dec 1986), and a B.Tech.
in Electrical Engineering from Indian Institute of Technology Madras (June 1984). The Company believes Mr. Rao is capable of making valuable
contributions to the Board due to his 15 years of experience as an executive, along with 25 years of entrepreneurial experience, including
in the biotech industry.
Lori
McNeill
Lori
McNeill has served on our Board since September 2022 and as Chairperson of our Business Advisory Committee since September 2019. Ms.
McNeill is the founder and Chief Executive Officer of McNeill Consulting, LLC since 2016, a management consulting company focused on
developing leaders to be more effective and ensuring that change management initiatives are seamless. Ms. McNeill has over twenty years’
experience in the healthcare industry, thirteen of which were at Pfizer Inc., which included working as the Chief of Staff of Global
Operations in the Integrated Health Business unit. From 2020 to 2021, Ms. McNeill was the Chief Operating Officer and Chairperson of
the board of directors of Global PPE, Inc., a worldwide supplier of personal protective equipment and safety supplies focused on healthcare
and government entities to fight the COVID-19 pandemic. She has been recognized by several institutions: Top 100 Global Women in Leadership
- Global Council for the Promotion of International Trade, 2021; Changemakers Summit Award Winner, 2021; The State of Women in Leadership
- Cover article for HR.com, 2020; and Pfizer International Innovation Excellence Award, 2011 and is currently Global Chairperson of Womenomics.
The Company believes Ms. McNeill is capable of making valuable contributions to the Board due to her over 20 years of experience in the
healthcare industry, including in senior leadership positions.
Executive
Officers
The
following table sets forth certain information about the current executive officers of the Company:
| Executive
Officers |
|
Age |
|
Position
and Office |
| Pankaj
Mohan, Ph.D. |
|
59 |
|
President
and Chief Executive Officer |
| Jay
Cross |
|
53 |
|
Chief
Financial Officer |
| John
K. Cini, Ph.D. |
|
71 |
|
Chief
Scientific Officer |
| Susan
Dexter |
|
68 |
|
Chief
Technical Officer |
| Richard
Kenney, M.D. |
|
65 |
|
Chief
Medical Officer |
Set
forth below are brief biographical descriptions of the individuals currently serving as the Company’s executive officers, based
on information furnished to the Company by such individuals.
Pankaj
Mohan, Ph.D.
See
description under Directors.
Jay
Cross
Jay
Cross joined Sonnet in May 2019 and has since served as its Chief Financial Officer and Chief Business Officer, and was appointed Chief
Financial Officer of the Company at the closing of the Merger. Prior to Sonnet, Mr. Cross was a Managing Director with Chardan Capital’s
healthcare investment banking team from November 2015 to March 2019, where he focused on biopharmaceuticals. Prior to that, from May
2014 to June 2015, Mr. Cross served as a Director with Alere Financial Partners and from May 2011 to October 2013 as a Senior Analyst
at Balyasny Asset Management. He launched his career in finance in 1999 as an associate analyst covering biotechnology on the healthcare
equity research team at Hambrecht & Quist. Mr. Cross earned an M.P.H. from the Yale University School of Medicine and a B.S. in psychology
from Washington & Lee University.
John
K. Cini, Ph.D.
John
K. Cini, Ph.D. co-founded Sonnet in 2015 and has since served as its Chief Scientific Officer, and was appointed Chief Scientific Officer
of the Company at the closing of the Merger, where he oversees and directs the Company’s discovery and development programs. His
role includes the oversight of the selection process of cancer and immune oncology targets and proof-of-concept testing. Prior to joining
Sonnet, he was Vice President of Discovery and Development Sciences at Oncobiologics, Inc. from January 2011 to April 2015. Dr. Cini
has successfully advanced more than 20 novel monoclonal antibody products from discovery to IND. He is the holder of several novel product
and formulation patents and applications. He has also been directly involved in several successful novel biologics through early discovery
research into development and manufacturing through clinical trials and commercialization. Previous positions include Executive Director
at Mederex (acquired by Bristol-Myers Squibb in 2010), lead discovery scientific roles at Johnson & Johnson (Ethicon, OrthoBioTech
& Pharmaceutical Research), and Bayer. Dr. Cini’s therapeutic areas of expertise in system biology include oncology, immune
oncology, inflammation, osteoporosis, wound healing, surgical adhesion and cellular aging. Dr. Cini has a PhD in Biochemistry from University
of North Texas.
Susan
Dexter
Susan
Dexter has served as a contract consultant to Sonnet in the capacity of Chief Technical Officer since May 2019 and was appointed full-time
Chief Technical Officer of the Company at the closing of the Merger on April 1, 2020. Her role at Sonnet is to manage the operations
for drug development from cell line development, through cGMP manufacturing of drug substance and clinical drug product, regulatory submissions
to initiate human clinical trials, and supply chain for labeling/packaging and distribution to clinical depots. All activities fall under
the FDA’s Chemical Manufacturing and Controls for biological drugs (“CMC”). She is also responsible for drug supply
and management of non-clinical animal studies in support of regulatory filings related to first-in-human studies. She came to Sonnet
with more than thirty years of experience in biotechnology science, manufacturing and business development, having been directly involved
in three start-up companies and multiple M&A activities. Her expertise in CMC for biologics process development ranges from cell
line development to process development through commercial manufacturing. In her role as Managing Director at Latham Biopharm Group (“LBG”)
from September 2008 until the closing of the Merger, Ms. Dexter ran the Product Development service offering, managing the activities
and disciplines related to pre- clinical toxicology studies, and CMC-related activities including IND filings, Quality oversight of cGMP
activities and other related CMC supply chain activities. She came to LBG from Xcellerex, Inc., a CDMO and developer of single use technology
for bioprocessing. She was Chief Business Officer at Xcellerex from April 2004 to September 2008. Prior to Xcellerex, from July 1998
to April 2004, she was VP of Business Development at The Dow Chemical Company’s CDMO, an acquisition of Collaborative BioAlliance,
facilitated by Ms. Dexter in 2000, and Assoc. Director of Business Development at Celltech Biologics, purchased by Lonza Biologics, a
biologics CDMO. She worked at Celltech/Lonza from 1986 to July 1998. Ms. Dexter holds a double major with Honors in Immunology and Marketing
from American University, Washington, D.C., and certifications from Harvard University in ‘Negotiations for Lawyers’ and
‘Finance for Non-financial Managers’. She was also Professor Emeritus at University College, London, Department of Bioengineering,
teaching a credited course lecture and workshop in “Project managing of a biologics facility”, to graduate, Ph.D. and post-graduate
professionals, from 1999 to 2006. She has served as a non-executive board member for Sartorius Stedim Biotech since 2015, compensation
committee member since 2019, and auditing committee member since 2022. In February 2023, Susan was appointed to the board of directors
for a London, UK based company Virocell, a technology developer and CDMO for manufacture of viral vectors for cell and gene therapies.
Richard
Kenney, M.D.
Richard
Kenney, M.D. has served as the Company’s Chief Medical Officer since April 2021. Dr. Kenney has more than 20 years of experience
in translational-stage development of biologics, as well as the commercialization strategy and corporate management of preclinical, clinical-stage
and commercialized vaccines and immunotherapies. As President of ClinReg Biologics, he has provided strategic consulting in clinical
and regulatory affairs of biologics and medical monitoring and pharmacovigilance in several capacities. As such, Dr. Kenney also serves
as the CMO for Public Health Vaccines’ Marburg vaccine program. He previously served as Chief Development Officer at X-VAX Technology
and before that had Chief Medical Officer roles at Immune Design and Crucell Holland, where he led the clinical development and regulatory
affairs groups. Dr. Kenney was a researcher/reviewer for the FDA for over six years and did post-graduate training at Duke and NIH. Dr.
Kenney received a B.S. in Chemistry from George Washington University and his M.D. from Harvard Medical School.
Section
16(a) Beneficial Ownership Reporting Compliance
Section
16(a) of the Securities Exchange Act of 1934, as amended, requires the Company’s directors and executive, officers, and persons
who are beneficial owners of more than 10% of a registered class of the Company’s equity securities, to file reports of ownership
and changes in ownership with the SEC. These persons are required by SEC regulations to furnish the Company with copies of all Section
16(a) forms they file.
Based
solely upon the Company’s review of copies of Forms 3, 4 and 5 furnished to the Company, the Company believes that all of its directors,
executive officers and any other applicable stockholders timely filed all reports required by Section 16(a) of the Exchange Act during
the fiscal year ended September 30, 2023, except for the following: Form 4s that were due on December 16, 2022 were filed on December 22, 2022 for Nailesh Bhatt, Albert Dyrness, Raghu
Rao, Lori McNeil, Pankaj Mohan, John Cini, Jay Cross, Susan Dexter and Rick Kenney; Form 4s that were due on January 10, 2023 were filed
on February 17, 2023 for Pankaj Mohan, John Cini, Susan Dexter and Rick Kenney; and a Form 4 that was due on December 13, 2022 was filed
on February 17, 2023 for Donald Griffith.
Code
of Business Conduct and Ethics
The
Company has adopted a Code of Business Conduct and Ethics that applies to its directors, officers and employees. The purpose of the Code
of Business Conduct and Ethics is to deter wrongdoing and to provide guidance to the Company’s directors, officers and employees
to help them recognize and deal with ethical issues, to provide mechanisms to report unethical or illegal conduct and to contribute positively
to the Company’s culture of honesty and accountability. The Company’s Code of Business Conduct and Ethics is publicly available
on the Company’s website at https://www.sonnetbio.com/. If the Company makes any substantive amendments to the Code of Business
Conduct and Ethics or grants any waiver, including any implicit waiver from a provision of the Code of Business Conduct and Ethics to
its directors or executive officers, the Company will disclose the nature of such amendments or waiver on its website or in a current
report on Form 8-K.
Audit
Committee
The
Board has established an Audit Committee currently consisting of Messrs. Bhatt (Chairman), Dyrness and Rao. The Audit Committee’s
primary functions are to oversee and review: the integrity of the Company’s consolidated financial statements and other financial
information furnished by the Company, the Company’s compliance with legal and regulatory requirements, the Company’s systems
of internal accounting and financial controls, the independent auditor’s engagement, qualifications, performance, compensation
and independence, related party transactions, and compliance with the Company’s Code of Business Conduct and Ethics.
Each
member of the Audit Committee is “independent” as that term is defined under the applicable rules of the SEC and the applicable
rules of The Nasdaq Stock Market. The Board has determined that each Audit Committee member has sufficient knowledge in financial and
auditing matters to serve on the Committee. The Board determined that Mr. Rao is an “audit committee financial expert,” as
defined under the applicable rules of the SEC and the applicable rules of The Nasdaq Stock Market. The Company’s Board has adopted
an Audit Committee Charter, which is available for viewing at https://www.sonnetbio.com/.
Item
11. Executive Compensation.
Summary
Compensation Table
The
following table shows the compensation awarded to or earned by each person serving as the Company’s principal executive officer
during fiscal year 2023, the Company’s two most highly compensated executive officers who were serving as executive officers as
of September 30, 2023 and up to two additional individuals for whom disclosure would have been provided but for the fact that such individuals
were not serving as an executive officer as of September 30, 2023. The persons listed in the following table are referred to herein as
the “named executive officers.”
SUMMARY
COMPENSATION TABLE
| Name
and Principal Position | |
Year | | |
Salary ($) | | |
Bonus ($) | | |
Stock awards ($)(1) | | |
Option awards ($)(1) | | |
All
other compensation ($) | | |
Total
($) | |
| Pankaj
Mohan, Ph.D. | |
2023 | | |
| 538,998 | | |
| 309,924 | | |
| 95,724 | | |
| - | | |
| 78,074 | | |
| 1,022,720 | |
| President
and Chief Executive Officer | |
2022 | | |
| 559,729 | | |
| 218,680 | | |
| 144,061 | | |
| - | | |
| 82,923 | | |
| 1,005,393 | |
| John
Cini, Ph.D. | |
2023 | | |
| 417,750 | | |
| 146,490 | | |
| 23,931 | | |
| - | | |
| 38,240 | | |
| 626,411 | |
| Chief
Scientific Officer | |
2022 | | |
| 413,048 | | |
| 113,899 | | |
| 36,015 | | |
| - | | |
| 52,014 | | |
| 614,976 | |
| Jay
Cross | |
2023 | | |
| 388,725 | | |
| 178,813 | | |
| 15,829 | | |
| - | | |
| 35,882 | | |
| 619,250 | |
| Chief
Financial Officer | |
2022 | | |
| 403,676 | | |
| 127,649 | | |
| 30,128 | | |
| - | | |
| 43,358 | | |
| 604,811 | |
| (1) |
Represents
the aggregate grant date fair value for grants made in 2023 and 2022 computed in accordance with Financial Accounting Standards Board
(“FASB”) Accounting Standards Codification (“ASC”) Topic 718. This calculation does not give effect to any
estimate of forfeitures related to service-based vesting, but assumes that the executive will perform the requisite service for the
award to vest in full. |
Narrative
Disclosure to Summary Compensation Table
Employment
Agreements
The
material terms of each named executive officer’s employment agreement or arrangement are described below.
Sonnet
entered into an employment agreement with Dr. Mohan on December 31, 2018, as amended (the “Mohan Agreement”), setting forth
the terms of his employment as Chief Executive Officer, which agreement was assumed by the Company at the closing of the Merger. Pursuant
to the employment agreement, Dr. Mohan is entitled to, among other things, (i) an annual gross base salary of $490,000, (ii) eligibility
for a bonus equal to 5.4% of gross revenue received by the Company from a strategic transaction and (iii) for any year in which the bonus
in the previous clause amounts to less than 50% of the base salary, an additional performance-based cash bonus to bring the aggregate
cash bonus for such year to up to 50% of the base salary, as determined by the board. The employment agreement shall terminate in accordance
with its terms. Pursuant to Dr. Mohan’s employment agreement, if he is terminated without “Cause” or for “Good
Reason” within 2 months prior to or within 12 months following a “Change in Control”, he is entitled to (i) his base
salary for 18 months, (ii) a bonus equal to his performance bonus for the year in which the termination occurs, divided by 12, and then
multiplied by 18, and (iii) if he timely continued coverage under COBRA, payment for COBRA premiums necessary to continue coverage until
the earliest of (a) 18 months following the termination date, (b) the date he becomes eligible for substantially equivalent health insurance
coverage in connection with new employment or self-employment, or (c) the date he becomes ineligible for COBRA continuation coverage.
If Dr. Mohan is terminated without “Cause” or for “Good Reason” not coincident with a “Change in Control”,
he is entitled to (i) his base salary for 18 months, (ii) any performance bonus for the performance year in which his termination occurs,
and (iii) if he timely continued coverage under COBRA, payment for COBRA premiums necessary to continue coverage until the earliest of
(a) 18 months following the termination date, (b) the date he becomes eligible for substantially equivalent health insurance coverage
in connection with new employment or self-employment, or (c) the date he becomes ineligible for COBRA continuation coverage.
Sonnet
entered into an employment agreement with Dr. Cini on January 10, 2020, as amended (the “Cini Agreement”), setting forth
the terms of his employment as Chief Scientific Officer, which agreement was assumed by the Company at the closing of the Merger. Pursuant
to the employment agreement, Dr. Cini is entitled to, among other things, (i) an annual gross base salary of $370,000, (ii) eligibility
for a bonus equal to 1.1% of gross revenue received by the Company from a strategic transaction and (iii) for any year in which the bonus
in the previous clause amounts to less than 35% of the base salary, an additional performance-based cash bonus to bring the aggregate
cash bonus for such year to up to 35% of the base salary, as determined by the board. The employment agreement shall terminate in accordance
with its terms. Pursuant to Dr. Cini’s employment agreement, if he is terminated without “Cause” or for “Good
Reason” within 2 months prior to or within 12 months following a “Change in Control”, he is entitled to (i) his base
salary for 12 months and (ii) if he timely continued coverage under COBRA, payment for COBRA premiums necessary to continue coverage
until the earliest of (a) 18 months following the termination date, (b) the date he becomes eligible for substantially equivalent health
insurance coverage in connection with new employment or self-employment, or (c) the date he becomes ineligible for COBRA continuation
coverage. If Dr. Cini is terminated without “Cause” or for “Good Reason” not coincident with a “Change
in Control”, he is entitled to (i) his base salary for 9 months and (ii) if he timely continued coverage under COBRA, payment for
COBRA premiums necessary to continue coverage until the earliest of (a) 12 months following the termination date, (b) the date he becomes
eligible for substantially equivalent health insurance coverage in connection with new employment or self-employment, or (c) the date
he becomes ineligible for COBRA continuation coverage.
Sonnet
entered into an employment agreement with Mr. Cross on January 10, 2020 (the “Cross Agreement”), setting forth the terms
of his employment as Chief Financial Officer, which agreement was assumed by the Company at the closing of the Merger. Pursuant to the
employment agreement, Mr. Cross is entitled to, among other things, (i) an annual gross base salary of $365,000 and (ii) eligibility
for a performance-based cash bonus of up to 40% of the base salary, as determined by the board. The employment agreement shall terminate
in accordance with its terms. Pursuant to Mr. Cross’s employment agreement, if he is terminated without “Cause” or
for “Good Reason” within 2 months prior to or within 12 months following a “Change in Control”, he is entitled
to (i) his base salary for 12 months, (ii) any performance bonus for the performance year in which his termination occurs, and (iii)
if he timely continued coverage under COBRA, payment for COBRA premiums necessary to continue coverage until the earliest of (a) 18 months
following the termination date, (b) the date he becomes eligible for substantially equivalent health insurance coverage in connection
with new employment or self-employment, or (c) the date he becomes ineligible for COBRA continuation coverage. If Mr. Cross is terminated
without “Cause” or for “Good Reason” not coincident with a “Change in Control”, he is entitled to
(i) his base salary for 9 months, (ii) any performance bonus for the performance year in which his termination occurs, and (iii) if he
timely continued coverage under COBRA, payment for COBRA premiums necessary to continue coverage until the earliest of (a) 12 months
following the termination date, (b) the date he becomes eligible for substantially equivalent health insurance coverage in connection
with new employment or self-employment, or (c) the date he becomes ineligible for COBRA continuation coverage.
Other
Agreements
On
April 1, 2020, the Company entered into an employment agreement with Ms. Dexter (the “Dexter Agreement”), setting forth the
terms of her employment as Chief Technical Officer. Pursuant to the employment agreement, Ms. Dexter is entitled to, among other things,
(i) an annual gross base salary of $310,000 and (ii) eligibility for a performance-based cash bonus of up to 35% of the base salary,
as determined by the board. The employment agreement shall terminate in accordance with its terms. Pursuant to Ms. Dexter’s employment
agreement, if she is terminated without “Cause” or for “Good Reason” within 2 months prior to or within 12 months
following a “Change in Control”, she is entitled to (i) her base salary for 12 months, (ii) any performance bonus for the
performance year in which her termination occurs, and (iii) if she timely continued coverage under COBRA, payment for COBRA premiums
necessary to continue coverage until the earliest of (a) 18 months following the termination date, (b) the date she becomes eligible
for substantially equivalent health insurance coverage in connection with new employment or self-employment, or (c) the date she becomes
ineligible for COBRA continuation coverage. If Ms. Dexter is terminated without “Cause” or for “Good Reason”
not coincident with a “Change in Control”, she is entitled to (i) his base salary for 9 months, (ii) any performance bonus
for the performance year in which her termination occurs, and (iii) if she timely continued coverage under COBRA, payment for COBRA premiums
necessary to continue coverage until the earliest of (a) 12 months following the termination date, (b) the date she becomes eligible
for substantially equivalent health insurance coverage in connection with new employment or self-employment, or (c) the date she becomes
ineligible for COBRA continuation coverage.
Outstanding
Equity Awards at Fiscal Year End
The
following table sets forth certain information, on an award-by-award basis, concerning unexercised options to purchase common stock,
restricted shares of common stock and common stock that has not yet vested for each named executive officer and outstanding as of September
30, 2023.
OUTSTANDING
EQUITY AWARDS AT FISCAL YEAR END - 2023
| | |
Stock
Awards | |
| Name | |
Equity
incentive plan awards: Number of unearned shares, units or other rights that have not vested (#) | | |
Equity
incentive plan awards: Market or payout value of unearned shares, units or other rights that have not vested ($) | |
| Pankaj
Mohan Ph.D. | |
| 3,343 | (1) | |
| 9,680 | |
| | |
| | | |
| | |
| John
Cini, Ph.D. | |
| 843 | (1) | |
| 2,420 | |
| | |
| | | |
| | |
| Jay
Cross | |
| 705 | (2) | |
| 2,025 | |
| (1) |
Each
restricted stock award vests 100% on January 1, 2024. |
| (2) |
Each
restricted stock unit (“RSU”) award vests 100% on January 1, 2024 |
Director
Compensation
Non-Employee
Director Compensation Policy
In
connection with the Merger, the Board approved a new director compensation policy for its non-employee directors. Other than reimbursement
for reasonable expenses incurred in connection with attending board and committee meetings, this policy provides for the following cash
compensation:
●
each non-employee director is entitled to receive an annual fee from us of $35,000;
●
the chair of our audit committee will receive an annual fee from us of $15,000;
●
the chair of our compensation committee will receive an annual fee from us of $10,000;
●
the chair of our nominating and corporate governance committee will receive an annual fee from us of $8,000; and
●
each non-chairperson member of the audit committee, the compensation committee and the nominating and corporate governance committee
will receive annual fees from us of $7,500, $5,000 and $4,000, respectively.
Each
non-employee director that joins the Board receives an initial option grant to purchase 0.080% of the Company’s fully-diluted outstanding
Common Stock at the closing of the Merger, which shall vest 33% per year over three years, the first vesting date to occur on the one-year
anniversary of the grant date. Each non-employee director also receives an annual option grant to purchase 0.040% of the Company’s
fully-diluted outstanding Common Stock at the closing of the Merger, which shall vest 100% upon the earlier of the one-year anniversary
of the grant date or the next annual stockholder meeting. Upon a change in control, as defined in the Company’s equity incentive
plan, 100% of the shares underlying these options shall become vested and exercisable immediately prior to such change in control.
Except
as set forth in the table below, the non-employee directors did not receive any cash or equity compensation during fiscal year 2023:
DIRECTOR
COMPENSATION
| Name | |
Fees
Earned or
Paid in
Cash ($) | | |
Stock Awards ($)(1) | | |
Option Awards ($)(1) | | |
All
Other Compensation ($) | | |
Total
($) | |
| Nailesh
Bhatt(2) | |
| 54,000 | | |
| 3,845 | | |
| - | | |
| - | | |
| 57,845 | |
| Albert
Dyrness(3) | |
| 55,500 | | |
| 3,845 | | |
| - | | |
| - | | |
| 59,345 | |
| Donald
Griffith (4) | |
| - | | |
| 3,278 | | |
| - | | |
| 134,048 | | |
| 137,326 | |
| Raghu
Rao(5) | |
| 116,500 | | |
| 3,845 | | |
| - | | |
| - | | |
| 120,345 | |
| Lori
McNeill(6) | |
| 70,000 | | |
| 3,845 | | |
| - | | |
| - | | |
| 73,845 | |
| (1) |
Represents
the aggregate grant date fair value for grants made in 2023 computed in accordance with FASB ASC Topic 718. This calculation does
not give effect to any estimate of forfeitures related to service-based vesting, but assumes that the executive will perform the
requisite service for the award to vest in full. |
| (2) |
Mr.
Bhatt holds an aggregate of 171 restricted stock units, as of September 30, 2023. |
| (3) |
Mr.
Dyrness holds an aggregate of 171 restricted stock units, as of September 30, 2023. |
| (4) |
Mr.
Griffith has served as Sonnet’s Financial Controller since January 1, 2019, and since the Merger serves as our Controller.
The amounts in the table above under “All Other Compensation” represent salary and bonus earned by Mr. Griffith for the
fiscal year 2023. See the description of the employment agreement with Mr. Griffith below. Mr. Griffith holds an aggregate of 146
restricted stock units, as of September 30, 2023. |
| (5) |
Mr.
Rao holds an aggregate of 171 restricted stock units, as of September 30, 2023. |
| (6) |
Ms.
McNeill holds an aggregate of 171 restricted stock units, as of September 30, 2023. |
Other
Agreement with a Director
Sonnet
entered into an employment agreement with Mr. Griffith on January 1, 2019, setting forth the terms of his employment as Financial Controller.
Pursuant to the employment agreement, Mr. Griffith is entitled to, among other things, (i) an annual prorated gross base salary of $150,000
and (ii) eligibility for a target bonus equal 25% of gross salary earned. The employment agreement has no specific term and constitutes
an at-will employment.
Item
12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
Security
Ownership of Certain Beneficial Owners and Management
The
following table sets forth certain information as of December 11, 2023 with respect to the beneficial ownership of common stock of the
Company by the following: (i) each of the Company’s current directors; (ii) each of the named executive officers; (iii) all of
the current executive officers and directors as a group; and (iv) each person known by the Company to own beneficially more than five
percent (5%) of the outstanding shares of the Company’s common stock.
For
purposes of the following table, beneficial ownership is determined in accordance with the applicable SEC rules and the information is
not necessarily indicative of beneficial ownership for any other purpose. Except as otherwise noted in the footnotes to the table, the
Company believes that each person or entity named in the table has sole voting and investment power with respect to all shares of the
Company’s common stock shown as beneficially owned by that person or entity (or shares such power with his or her spouse). Under
the SEC’s rules, shares of the Company’s common stock issuable under options that are exercisable on or within 60 days after
December 11, 2023 (“Presently Exercisable Options”) are deemed outstanding and therefore included in the number of shares
reported as beneficially owned by a person or entity named in the table and are used to compute the percentage of the common stock beneficially
owned by that person or entity. These shares are not, however, deemed outstanding for computing the percentage of the common stock beneficially
owned by any other person or entity.
The
percentage of the common stock beneficially owned by each person or entity named in the following table is based on 3,069,516 shares
of common stock issued and outstanding as of December 11, 2023 plus any shares issuable upon exercise of Presently Exercisable Options
held by such person or entity.
Title
of Class | |
Name
And Address of Beneficial Owner* | |
Amount
And Nature of Beneficial Ownership | | |
Percent
Of Class | |
| |
Named
Executive Officers and Directors: | |
| | | |
| | |
| |
Pankaj
Mohan, Ph.D. | |
| 131,322 | (1) | |
| 4.2 | % |
| |
Nailesh
Bhatt | |
| 1,396 | (2) | |
| ** | |
| | |
Albert
Dyrness | |
| 1,316 | (3) | |
| ** | |
| | |
Donald
Griffith | |
| 468 | (4) | |
| ** | |
| | |
Raghu
Rao | |
| 47,117 | (5) | |
| 1.5 | % |
| | |
Lori
McNeill | |
| 171 | (6) | |
| ** | |
| | |
John.
K. Cini, Ph.D. | |
| 1,830 | | |
| ** | |
| | |
Jay
Cross | |
| 1,087 | (7) | |
| ** | |
| | |
All
current executive officers and directors as a group (10 persons) | |
| 187,064 | | |
| 5.9 | % |
| | |
| |
| | | |
| | |
| | |
5%
Holders | |
| | | |
| | |
| | |
John
Markey | |
| 231,115 | | |
| 7.5 | % |
| (*) |
Unless
otherwise indicated, the address is c/o Sonnet BioTherapeutics, Inc., 100 Overlook Center, Suite 102, Princeton, New Jersey, 08540. |
| |
|
| (**) |
Less
than 1%. |
| |
|
| (1) |
Includes
(i) 3,021 shares of common stock held by the Mohan Family Office, over which Dr. Mohan has shared power to vote and dispose with
Swati Mohan, his spouse and (ii) 25 shares of common stock held individually by Pankhuri Mohan, Dr. Mohan’s child, over which
Dr. Mohan has shared power to vote and dispose with Pankhuri Mohan. Includes 68,750 shares of common stock currently issuable upon
the exercise of warrants. |
| (2) |
Includes
171 restricted stock units, which will be settled in shares of common stock and vest within 60 days of December 11, 2023. |
| |
|
| (3) |
Includes
171 restricted stock units, which will be settled in shares of common stock and vest within 60 days of December 11, 2023. |
| |
|
| (4) |
Includes
146 restricted stock units, which will be settled in shares of common stock and vest within 60 days of December 11, 2023. |
| |
|
| (5) |
Includes
31,250 shares of common stock issuable upon exercise of warrants which are exercisable within 60 days of December 11, 2023. Includes
171 restricted stock units, which will be settled in shares of common stock and vest within 60 days of December 11, 2023. |
| |
|
| (6) |
Includes
171 restricted stock units, which will be settled in shares of common stock and vest within 60 days of December 11, 2023. |
| |
|
| (7) |
Excludes
705 restricted stock units, which will be settled in shares of common stock and vest within 60 days of December 11, 2023. |
Equity
Compensation Plan Information
The
following table provides information as of September 30, 2023 regarding shares of the Company’s common stock that may be issued
under the Company’s existing equity compensation plans, including its 2020 Omnibus Equity Incentive Plan (the “2020 Plan”).
| | |
Equity
Compensation Plan Information | |
| Plan
Category | |
(a) Number
of securities to
be issued upon exercise of
outstanding options, warrants and
rights | | |
(b) Weighted
average exercise
price of outstanding
options, warrants and
rights | | |
(c) Number
of securities remaining
available for future
issuance under
equity compensation
plans (excluding
securities reflected
in column (a)) | |
| Equity
compensation plans approved by security holders (1) | |
| 2,326 | | |
| N/A | | |
| - | |
| Equity
compensation plans not approved by security holders | |
| N/A | | |
| N/A | | |
| N/A | |
| Total | |
| 2,326 | | |
| - | | |
| - | |
| (1) |
The
weighted-average exercise price does not reflect the shares that will be issued in connection with the settlement of RSUs, since
RSUs have no exercise price. Other than RSUs, there were no outstanding options, warrants, or rights under our equity compensation
plan as of September 30, 2023. |
Item
13. Certain Relationships and Related Transactions, and Director Independence.
Other
than compensation arrangements for named executive officers and directors, the Company describes below each transaction and series of
similar transactions, since the beginning of fiscal year 2022, to which the Company was a party or will be a party, in which:
| ● |
the
amounts involved exceeded or will exceed the lesser of $120,000 or one percent of the average of the smaller reporting company’s
total assets at year-end for the last two completed fiscal years; and |
| ● |
any
of the Company’s directors, nominees for director, executive officers or holders of more than 5% of the Company’s common
stock, or any member of the immediate family of the foregoing persons, had or will have a direct or indirect material interest. |
Compensation
arrangements for the Company’s named executive officers and directors are described in the section entitled “Executive Compensation”.
Public
Offering
Pankaj
Mohan, our Chairman and Chief Executive Officer, purchased 34,375 shares of common stock and 68,750 warrants to purchase 68,750 shares
of common stock pursuant to an underwritten public offering by the us at $1.60 per share and accompanying two warrants. The offering
closed on October 27, 2023.
Raghu
Rao, a director, purchased 15,625 shares of common stock and 31,250 warrants to purchase 31,250 shares of common stock pursuant to an
underwritten public offering by the us at $1.60 per share and accompanying two warrants. The offering closed on October 27, 2023.
Indemnification
Agreements
The
Company has entered into indemnification agreements with each of its current directors and executive officers. These agreements will
require the Company to indemnify these individuals to the fullest extent permitted under Delaware law against liabilities that may arise
by reason of their service to the Company, and to advance expenses incurred as a result of any proceeding against them as to which they
could be indemnified. The Company also intends to enter into indemnification agreements with its future directors and executive officers.
Director
Independence
The
Company is currently managed by a six-member board of directors. The Company has determined that Messrs. Bhatt, Dyrness and Rao and Ms.
McNeill are “independent” as that term is defined under the rules of The NASDAQ Stock Market.
Item
14. Principal Accountant Fees and Services.
The
following table summarizes the fees for professional services rendered by KPMG LLP, the Company’s (and Sonnet’s, prior to
the Merger) independent registered public accounting firms, for each of the respective last two fiscal years:
| Fee
Category | |
2023 | | |
2022 | |
| Audit
Fees | |
$ | 540,000 | | |
$ | 435,000 | |
| Audit-Related
Fees | |
| - | | |
| - | |
| Tax
Fees | |
| 61,565 | | |
| 33,500 | |
| All
Other Fees | |
| - | | |
| - | |
| Total
Fees | |
$ | 601,565 | | |
$ | 468,500 | |
Audit
Fees
Represents
fees for professional services provided in connection with the audit of the Company’s annual consolidated financial statements
and reviews of the Company’s quarterly interim consolidated financial statements, as well as for fees associated with registration
statements, comfort letters and consents.
Audit-Related
Fees
We
did not incur any audit-related fees from our independent auditors in 2023 or 2022.
Tax
Fees
Tax
fees are associated with tax compliance, tax advice, tax planning and tax preparation services.
All
Other Fees
Fees
related to products and services provided by the principal accountant, other than the services reported in the above sections.
The
Audit Committee is responsible for appointing, setting compensation and overseeing the work of the independent auditors. The Audit Committee
is required to review and approve the proposed retention of independent auditors to perform any proposed auditing and non-auditing services
as outlined in its charter. The Audit Committee has not established policies and procedures separate from its charter concerning the
pre-approval of auditing and non-auditing related services. As required by Section 10A of the Exchange Act, our Audit Committee has authorized
all auditing and non-auditing services provided by KPMG LLP during 2023 and 2022 and the fees paid for such services. However, the pre-approval
requirement may be waived with respect to the provision of non-audit services for the Company if the “de minimis” provisions
of Section 10A(i)(1)(B) of the Exchange Act are satisfied.
The
Audit Committee has considered whether the provision of Audit-Related Fees, Tax Fees, and all other fees as described above is compatible
with maintaining KPMG LLP’s independence and has determined that such services for fiscal years 2023 and 2022 were compatible.
All such services were approved by the Audit Committee pursuant to Rule 2-01 of Regulation S-X under the Exchange Act to the extent that
rule was applicable.
The
Audit Committee is responsible for reviewing and discussing the audited consolidated financial statements with management, discussing
with the independent registered public accountants the matters required by Public Company Accounting Oversight Board Auditing Standard
No. 1301 Communications with Audit Committees, receiving written disclosures from the independent registered public accountants
required by the applicable requirements of the Public Company Accounting Oversight Board regarding the independent registered public
accountants’ communications with the Audit Committee concerning independence and discussing with the independent registered public
accountants their independence, and recommending to the Board that the audited consolidated financial statements be included in the Company’s
Annual Report on Form 10-K.
PART
IV
Item
15. Exhibits, Financial Statement Schedules.
(a)(1)
Financial Statements. The financial statements filed as part of this report are listed on the Index to the Consolidated Financial
Statements.
(a)(2)
Financial Statement Schedules. Schedules are omitted because they are not applicable or the required information is shown in the
consolidated financial statements or notes thereto.
(a)(3)
Exhibits. Reference is made to the Exhibit Index. The exhibits are included, or incorporated by reference, in this annual report
on Form 10-K and are numbered in accordance with Item 601 of Regulation S-K.
Item
16. Form 10-K Summary.
None.
SIGNATURES
Pursuant
to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed
on its behalf by the undersigned, thereunto duly authorized.
| |
Sonnet
BioTherapeutics, Inc.
(Registrant) |
| |
|
| Date:
December 14, 2023 |
/s/
Pankaj Mohan, Ph.D |
| |
Pankaj
Mohan, Ph.D |
| |
President
and Chief Executive Officer
(Principal
Executive Officer and
duly
authorized signatory) |
| |
|
| Date:
December 14, 2023 |
/s/
Jay Cross |
| |
Jay
Cross |
| |
Chief
Financial Officer
(Principal
Financial and Accounting Officer) |
SIGNATURES
AND POWER OF ATTORNEY
KNOW
ALL BY THESE PRESENTS, that each person whose signature appears below hereby constitutes and appoints Pankaj Mohan, Ph.D. and Jay Cross,
and each of them, his true and lawful agent, proxy and attorney-in-fact, with full power of substitution and resubstitution, for him
and in his name, place and stead, in any and all capacities, to (i) act on, sign and file with the Securities and Exchange Commission
any and all amendments to this annual report on Form 10-K together with all schedules and exhibits thereto, (ii) act on, sign and file
such certificates, instruments, agreements and other documents as may be necessary or appropriate in connection therewith and, (iii)
take any and all actions which may be necessary or appropriate to be done, as fully for all intents and purposes as he might or could
do in person, hereby approving, ratifying and confirming all that such agent, proxy and attorney-in-fact or any of his substitutes may
lawfully do or cause to be done by virtue thereof.
Pursuant
to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons on behalf of the registrant
and in the capacities and on the dates indicated.
| Signature |
|
Title |
|
Date |
| |
|
|
|
|
| /s/
Pankaj Mohan, Ph.D |
|
President,
Chief Executive Officer and Chairman |
|
December
14, 2023 |
| Pankaj
Mohan, Ph.D |
|
(Principal
Executive Officer) |
|
|
| |
|
|
|
|
| /s/
Jay Cross |
|
Chief
Financial Officer |
|
December
14, 2023 |
| Jay
Cross |
|
(Principal
Financial and Accounting Officer) |
|
|
| |
|
|
|
|
| /s/
Albert Dyrness |
|
Director |
|
December
14, 2023 |
| Albert
Dyrness |
|
|
|
|
| |
|
|
|
| /s/
Nailesh Bhatt |
|
Director |
|
December
14, 2023 |
| Nailesh
Bhatt |
|
|
|
|
| |
|
|
|
|
| /s/
Raghu Rao |
|
Director |
|
December
14, 2023 |
| Raghu
Rao |
|
|
|
|
| |
|
|
|
| /s/
Donald Griffith |
|
Director |
|
December
14, 2023 |
| Donald
Griffith |
|
|
|
|
| |
|
|
|
|
| /s/
Lori McNeill |
|
Director |
|
December
14, 2023 |
| Lori
McNeill |
|
|
|
|
INDEX
TO EXHIBITS
Exhibit
No. |
|
Description |
| |
|
|
| 2.1 |
|
Agreement
and Plan of Merger, dated October 10, 2019, by and among the Company, Sonnet Sub. and Merger Sub (filed as Exhibit 2.1 to the Company’s
Current Report on Form 8-K as filed on October 11, 2019, and incorporated herein by reference).# |
| |
|
|
| 2.2 |
|
Amendment
No. 1 to Agreement and Plan of Merger, dated February 7, 2020, by and among the Company, Sonnet Sub and Merger Sub (filed as Exhibit
2.1 to the Company’s Current Report on Form 8-K as filed on February 7, 2020, and incorporated herein by reference). |
| |
|
|
| 2.3 |
|
Share
Exchange Agreement, between Sonnet BioTherapeutics, Inc. and Relief Therapeutics Holding SA, dated August 9, 2019 (incorporated by
reference to Exhibit 2.10 to the Company’s Registration Statement on Form S-4 filed with the SEC on November 27, 2019).# |
| |
|
|
| 3.1 |
|
Certificate
of Incorporation, as amended, of Sonnet BioTherapeutics Holdings, Inc. (incorporated by reference to Exhibit 3.1 to the Company’s
Annual Report on Form 10-K filed with the SEC on December 17, 2020). |
| |
|
|
| 3.2 |
|
Certificate
of Designation of Preferences, Rights and Limitations of Series 3 Preferred Stock (incorporated by reference to Exhibit 3.1 to our
Current Report on Form 8-K, filed with the SEC on August 15, 2022). |
| |
|
|
| 3.3 |
|
Certificate
of Designation of Preferences, Rights and Limitations of Series 4 Preferred Stock (incorporated by reference to Exhibit 3.2 to our
Current Report on Form 8-K, filed with the SEC on August 15, 2022). |
| |
|
|
| 3.4 |
|
Certificate
of Amendment of Certificate of Incorporation, as amended, of Sonnet BioTherapeutics Holdings, Inc., dated September 16, 2022 (incorporated
by reference to Exhibit 3.1 to our Current Report on Form 8-K, filed with the SEC on September 19, 2022). |
| |
|
|
| 3.5 |
|
Certificate
of Amendment of Certificate of Incorporation, as amended, of Sonnet BioTherapeutics Holdings, Inc., dated August 31, 2023 (incorporated
by reference to Exhibit 3.1 to our Current Report on Form 8-K, filed with the SEC on September 1, 2023). |
| |
|
|
| 3.6 |
|
Amended
and Restated Bylaws, of Sonnet BioTherapeutics Holdings, Inc. (incorporated by reference to Exhibit 3.3 to our Current Report on
Form 8-K, filed with the SEC on August 15, 2022). |
| |
|
|
| 4.1 |
|
Form
of Common Stock Certificate (Incorporated by reference to Exhibit 4.1 to our Registration Statement on Form S-1 (Registration No.
333-178307), filed with the SEC on December 2, 2011). |
| |
|
|
| 4.2 |
|
Form
of Warrant dated May 4, 2017 (incorporated by reference to Exhibit 4.2 to our Current Report on Form 8-K, filed with the SEC on May
5, 2017). |
| |
|
|
| 4.3 |
|
Spin-Off
Entity Warrant, dated April 1, 2020 (incorporated by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed
with the SEC on April 3, 2020). |
| |
|
|
| 4.4 |
|
Form
of Sonnet BioTherapeutics, Inc. Converted Warrant (incorporated by reference to Exhibit 4.2 to the Company’s Quarterly Report
on Form 10-Q filed with the SEC on August 14, 2020). |
| |
|
|
| 4.5 |
|
Form
of Series A/B Warrants (incorporated by reference to Exhibit 4.16 to the Company’s Registration Statement on Form S-4/A filed
with the SEC on February 7, 2020). |
| |
|
|
| 4.6 |
|
Form
of Series C Warrant (incorporated by reference to Exhibit 4.1 to our Current Report on Form 8-K filed with the SEC on August 4, 2020). |
| |
|
|
| 4.7 |
|
Registration
Rights Agreement, dated February 7, 2020, by and between the Company and certain investors named therein (incorporated by reference
to Exhibit 4.17 to the Company’s Registration Statement on Form S-4/A filed with the SEC on February 7, 2020). |
| 4.8 |
|
Description
of Securities* |
| |
|
|
| 4.9 |
|
Form
of Pre-Funded Warrant, dated August 24, 2021 (incorporated by reference to Exhibit 4.2 to the Company’s Current Report on Form
8-K filed with the SEC August 25, 2021). |
| |
|
|
| 4.10 |
|
Form
of Underwriter Warrant, dated August 24, 2021 (incorporated by reference to Exhibit 4.9 to the Company’s Registration Statement
on Form S-1/A filed with the SEC on August 16, 2021). |
| |
|
|
| 4.11 |
|
Form
of Common Warrant, dated August 24, 2021 (incorporated by reference to Exhibit 4.4 to the Company’s Current Report on Form
8-K filed with the SEC August 25, 2021). |
| |
|
|
| 4.12 |
|
Form
of Pre-Funded Warrant dated February 10, 2023 (incorporated by reference to Exhibit 4.2 to our Current Report on Form 8-K filed with
the SEC on February 13, 2023). |
| |
|
|
| 4.13 |
|
Form
of Underwriter Warrant dated February 10, 2023 (incorporated by reference to Exhibit 4.3 to our Current Report on Form 8-K filed
with the SEC on February 13, 2023). |
| |
|
|
| 4.14 |
|
Form
of Common Warrant dated February 10, 2023 (incorporated by reference to Exhibit 4.4 to our Current Report on Form 8-K filed with
the SEC on February 13, 2023). |
| |
|
|
| 4.15 |
|
Form
of Pre-Funded Warrant dated June 30, 2023 (incorporated by reference to Exhibit 4.1 to Current Report on Form 8-K, filed with the
SEC on June 30, 2023). |
| |
|
|
| 4.16 |
|
Form
of Warrant dated June 30, 2023 (incorporated by reference to Exhibit 4.2 to Current Report on Form 8-K, filed with the SEC on June
30, 2023). |
| |
|
|
| 4.17 |
|
Form
of Placement Agent Warrant dated June 30, 2023 (incorporated by reference to Exhibit 4.3 to Current Report on Form 8-K, filed with
the SEC on June 30, 2023). |
| |
|
|
| 10.1 |
|
Common
Stock Purchase Agreement, between GEM Global Yield Fund LLC SCS and Sonnet BioTherapeutics, Inc., dated August 6, 2019 (incorporated
by reference to Exhibit 10.54 to the Company’s Registration Statement on Form S-4 filed with the SEC on November 27, 2019). |
| |
|
|
| 10.2 |
|
Amendment
to Common Stock Purchase Agreement, between GEM Global Yield Fund LLC SCS and Sonnet BioTherapeutics, Inc., dated September 25, 2019
(incorporated by reference to Exhibit 10.55 to the Company’s Registration Statement on Form S-4 filed with the SEC on November
27, 2019). |
| |
|
|
| 10.3 |
|
Side
Letter and Amendment No. 2 to Common Stock Purchase Agreement, between GEM Global Yield Fund LLC SCS, Sonnet BioTherapeutics, Inc.
and Chanticleer Holdings, Inc., dated February 7, 2020 (incorporated by reference to Exhibit 10.60 to the Company’s Registration
Statement on Form S-4 filed with the SEC on February 7, 2020). |
| |
|
|
| 10.4 |
|
Employment
Agreement, between Pankaj Mohan and Sonnet BioTherapeutics, Inc., dated December 31, 2018 (incorporated by reference to Exhibit 10.56
to the Company’s Registration Statement on Form S-4 filed with the SEC on February 7, 2020). † |
| |
|
|
| 10.5 |
|
Employment
Agreement, between John Cini and Sonnet BioTherapeutics, Inc., dated January 10, 2020 (incorporated by reference to Exhibit 10.58
to the Company’s Registration Statement on Form S-4 filed with the SEC on February 7, 2020). † |
| |
|
|
| 10.6 |
|
Employment
Agreement, between Jay Cross and Sonnet BioTherapeutics, Inc., dated January 10, 2020 (incorporated by reference to Exhibit 10.57
to the Company’s Registration Statement on Form S-4 filed with the SEC on February 7, 2020). † |
| |
|
|
| 10.7 |
|
Employment
Agreement, between Susan Dexter and the Company, dated April 1, 2020 (incorporated by reference to Exhibit 10.7 to the Company’s
Current Report on Form 8-K filed with the SEC on April 3, 2020). † |
| 10.8 |
|
Offer
Letter, between Donald Griffith and Sonnet BioTherapeutics, Inc., dated January 1, 2019 (incorporated by reference to Exhibit 10.59
to the Company’s Registration Statement on Form S-4 filed with the SEC on February 7, 2020). † |
| |
|
|
| 10.9 |
|
Sonnet
BioTherapeutics Holdings, Inc. 2020 Omnibus Equity Incentive Plan (incorporated by reference to Exhibit 4.2 to the Company’s
Registration Statement on Form S-8 filed with the SEC on May 20, 2020). † |
| |
|
|
| 10.10 |
|
Form
of Restricted Stock Unit Award (incorporated by reference to Exhibit 10.1 to our Current Report on Form 8-K (file No. 001-35570),
filed with the SEC on July 9, 2020). † |
| |
|
|
| 10.11 |
|
License
Agreement, between Ares Trading SA and Relief Therapeutics SA, dated August 28, 2015 (incorporated by reference to Exhibit 10.51
to the Company’s Registration Statement on Form S-4 filed with the SEC on February 7, 2020).*** |
| |
|
|
| 10.12 |
|
Discovery
Collaboration Agreement, between XOMA (US) LLC and Oncobiologics, Inc., dated July 23, 2012 (incorporated by reference to Exhibit
10.52 to the Company’s Registration Statement on Form S-4 filed with the SEC on February 7, 2020).*** |
| |
|
|
| 10.13 |
|
Amendment
of Discovery Collaboration Agreement, between XOMA (US) LLC and Sonnet BioTherapeutics, Inc., dated May 7, 2019 (incorporated by
reference to Exhibit 10.53 to the Company’s Registration Statement on Form S-4 filed with the SEC on February 7, 2020).*** |
| |
|
|
| 10.14 |
|
Securities
Purchase Agreement, dated as of February 7, 2020, by and among Chanticleer Holdings, Inc., Sonnet BioTherapeutics, Inc. and the investors
party thereto (incorporated by reference to Exhibit 10.64 to the Company’s Registration Statement on Form S-4 filed with the
SEC on February 7, 2020). |
| |
|
|
| 10.15 |
|
Form
of Warrant Exercise and Omnibus Amendment Agreement, dated as of August 3, 2020, by and between Sonnet BioTherapeutics Holdings,
Inc. and the Holders (incorporated by reference to Exhibit 10.1 to our Current Report on Form 8-K (File No. 001-35570), filed with
the SEC on August 4, 2020). |
| |
|
|
| 10.16 |
|
Assignment
and Assumption Employment Agreements by Sonnet BioTherapeutics Holdings, Inc., effective April 1, 2020 (incorporated by reference
to Exhibit 10.16 to the Company’s Annual Report on Form 10-K filed with the SEC on December 17, 2020).† |
| |
|
|
| 10.17 |
|
Amendment
No. 1 to Executive Employment Agreement, between Pankaj Mohan and the Company, dated November 23, 2020 (incorporated by reference
to Exhibit 10.17 to the Company’s Annual Report on Form 10-K filed with the SEC on December 17, 2020).† |
| |
|
|
| 10.18 |
|
Amendment
No. 1 to Executive Employment Agreement, between John Cini and the Company, dated November 23, 2020 (incorporated by reference to
Exhibit 10.18 to the Company’s Annual Report on Form 10-K filed with the SEC on December 17, 2020).† |
| |
|
|
| 10.19 |
|
Form
of Indemnification Agreement (incorporated by reference to Exhibit 10.19 to the Company’s Annual Report on Form 10-K filed
with the SEC on December 17, 2020).† |
| |
|
|
| 10.20 |
|
At-The-Market
Sales Agreement, dated February 5, 2020, between the Company and BTIG (incorporated by reference to Exhibit 1.1 to our Current Report
on Form 8-K (File No. 001-35570), filed with the SEC on February 5, 2021). |
| |
|
|
| 10.21 |
|
License
Agreement, dated May 2, 2021, between Sonnet BioTherapeutics, Inc. and New Life Therapeutics PTE, LTD (incorporated by reference
to Exhibit 10.1 to our Quarterly Report on Form 10-Q, filed with the SEC on May 17, 2021). |
| |
|
|
| 10.22 |
|
First
Amendment to License Agreement, dated June 11, 2021, between Sonnet BioTherapeutics, Inc. and New Life Therapeutics PTE, LTD (incorporated
by reference to Exhibit 10.22 to our Annual Report on Form 10-K, filed with the SEC on December 17, 2021). |
| |
|
|
| 10.23 |
|
Second
Amendment to License Agreement, dated July 7, 2021, among Sonnet Biotherapeutics CH SA, Sonnet BioTherapeutics, Inc. and New Life
Therapeutics PTE, Ltd. (incorporated by reference to Exhibit 10.23 to our Annual Report on Form 10-K, filed with the SEC on December
17, 2021). |
| |
|
|
10.24
|
|
Amendment
to License Agreement and Settlement, dated November 1, 2021, between ARES TRADING SA and Sonnet BioTherapeutics CH SA (incorporated
by reference to Exhibit 10.24 to our Annual Report on Form 10-K, filed with the SEC on December 17, 2021). |
| 10.25 |
|
At-The-Market
Sales Agreement, dated August 15, 2022, between the Company and BTIG (incorporated by reference to Exhibit 1.1 to our Current Report
on Form 8-K, filed with the SEC on August 15, 2022). |
| |
|
|
| 10.26 |
|
Underwriting
Agreement, dated February 8, 2023, between the Company and Chardan Capital Markets, LLC (incorporated by reference to Exhibit 1.1
to our Current Report on Form 8-K filed with the SEC on February 13, 2023). |
| |
|
|
| 10.27 |
|
Form
of Securities Purchase Agreement, dated June 28, 2023, by and between the Company and the
Purchaser (incorporated by reference to Exhibit 10.1 to our Current Report on Form 8-K filed
with the SEC on June 30, 2023).
|
| |
|
|
| 21.1 |
|
Subsidiaries
of the Company.* |
| |
|
|
| 23.1 |
|
Consent
of KPMG LLP.* |
| |
|
|
| 24.1 |
|
Power
of attorney (included on the signature page).* |
| |
|
|
| 31.1 |
|
Certification
of Principal Executive Officer pursuant to Rules 13a-14(a) and 15d-14(a) promulgated under the Securities and Exchange Act of 1934,
as amended.* |
| |
|
|
| 31.2 |
|
Certification
of Principal Financial Officer pursuant to Rules 13a-14(a) and 15d-14(a) promulgated under the Securities and Exchange Act of 1934,
as amended.* |
| |
|
|
| 32.1 |
|
Certification
of Principal Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of
2002.** |
| |
|
|
| 32.2 |
|
Certification
of Principal Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of
2002.** |
| |
|
|
| 97.1 |
|
Clawback Policy* |
| |
|
|
| 101.INS |
|
Inline
XBRL Instance Document - the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within
the Inline XBRL document.* |
| |
|
|
| 101.SCH |
|
Inline
XBRL Taxonomy Extension Schema Document.* |
| |
|
|
| 101.CAL |
|
Inline
XBRL Taxonomy Extension Calculation Linkbase Document.* |
| |
|
|
| 101.DEF |
|
Inline
XBRL Taxonomy Extension Definition Linkbase Document.* |
| |
|
|
| 101.LAB |
|
Inline
XBRL Taxonomy Extension Label Linkbase Document.* |
| |
|
|
| 101.PRE |
|
Inline
XBRL Taxonomy Extension Presentation Linkbase Document.* |
| |
|
|
| 104 |
|
Cover
Page Interactive Data File (formatted as inline XBRL and contained in Exhibit 101).* |
| * |
Filed
herewith. |
| ** |
Furnished
herewith. |
| *** |
Filed
herewith; portions of the exhibit have been omitted pursuant to Item 601(b)(10) of Regulation S-K. A copy of any omitted portions
will be furnished to the Securities and Exchange Commission upon request. |
| # |
The
schedules and exhibits to this agreement have been omitted pursuant to Item 601(b)(2) of Regulation S-K. A copy of any omitted schedule
and/or exhibit will be furnished to the Securities and Exchange Commission upon request. |
| † |
Indicates
a management contract or compensation plan, contract or arrangement. |
Exhibit
4.8
DESCRIPTION
OF SONNET BIOTHERAPEUTICS HOLDINGS, INC.’S SECURITIES
REGISTERED
PURSUANT TO SECTION 12 OF THE
SECURITIES
EXCHANGE ACT OF 1934
As
of September 30, 2023, Sonnet BioTherapeutics Holdings, Inc. (the “Company”) had two classes of securities registered under
Section 12 of the Securities Exchange Act of 1934, as amended: our voting common stock, $0.0001 par value per share and preferred stock,
par value $0.0001 per share.
DESCRIPTION
OF CAPITAL STOCK
The
following is a summary of information concerning capital stock of Sonnet BioTherapeutics Holdings, Inc. (“us,” “our,”
“we” or the “Company”) and does not purport to be complete. The summary is subject to, and qualified in its entirety
by reference to, Sonnet BioTherapeutics Holdings, Inc.’s certificate of incorporation, as amended, bylaws and the Delaware General
Corporation Law (the “DGCL”). You are urged to read our fourth amended and restated certificate of incorporation, as amended,
amended and restated bylaws and the applicable provisions of the DGCL for additional information.
General
Our
authorized capital stock consists of:
| |
● |
125,000,000
shares of common stock, par value $0.0001 per share; and |
| |
|
|
| |
● |
5,000,000
shares of preferred stock, par value $0.0001 per share, of which, as of the date of this report, none of which shares have been designated. |
As
of close of business on December 11, 2023, 3,069,516 shares of common stock were issued and outstanding and no shares of preferred stock
were issued and outstanding.
The
additional shares of our authorized stock available for issuance may be issued at times and under circumstances so as to have a dilutive
effect on earnings per share and on the equity ownership of the holders of our Common Stock. The ability of our board of directors to
issue additional shares of stock could enhance the board’s ability to negotiate on behalf of the stockholders in a takeover situation
but could also be used by the board to make a change-in-control more difficult, thereby denying stockholders the potential to sell their
shares at a premium and entrenching current management. The following description is a summary of the material provisions of our capital
stock. You should refer to our certificate of incorporation, as amended and bylaws, both of which are on file with the SEC as exhibits
to previous SEC filings, for additional information. The summary below is qualified by provisions of applicable law.
Common
Stock
Holders
of our Common Stock are each entitled to cast one vote for each share held of record on all matters presented to stockholders. Cumulative
voting is not allowed; the holders of a majority of our outstanding shares of Common Stock may elect all directors. Holders of our Common
Stock are entitled to receive such dividends as may be declared by our board out of funds legally available and, in the event of liquidation,
to share pro rata in any distribution of our assets after payment of liabilities. Our directors are not obligated to declare a dividend.
It is not anticipated that we will pau dividends in the foreseeable future. Holders of our do not have preemptive rights to subscribe
to any additional shares we may issue in the future. There are no conversion, redemption, sinking fund or similar provisions regarding
the Common Stock. All outstanding shares of Common Stock are fully paid and nonassessable.
The
rights, preferences and privileges of holders of Common Stock are subject to the rights of the holders of any outstanding shares of preferred
stock.
Transfer
Agent and Registrar
The
transfer agent and registrar for our Common Stock is Securities Transfer Corporation. The transfer agent address is Securities Transfer
Corporation, 2901 N Dallas Parkway, Suite 380, Plano, TX 75093, (469) 633-0101.
Preferred
Stock
We
are authorized to issue up to 5,000,000 shares of preferred stock, all of which are undesignated. Our board of directors has the authority
to issue preferred stock in one or more classes or series and to fix the designations, powers, preferences and rights, and the qualifications,
limitations or restrictions thereof, including dividend rights, conversion right, voting rights, terms of redemption, liquidation preferences
and the number of shares constituting any class or series, without further vote or action by the stockholders. Although we have no present
plans to issue any other shares of preferred stock, the issuance of shares of preferred stock, or the issuance of rights to purchase
such shares, could decrease the amount of earnings and assets available for distribution to the holders of common stock, could adversely
affect the rights and powers, including voting rights, of the common stock, and could have the effect of delaying, deterring or preventing
a change of control of us or an unsolicited acquisition proposal. The preferred stock may provide for an adjustment of the conversion
price in the event of an issuance or deemed issuance at a price less than the applicable conversion price, subject to certain exceptions.
If
we offer a specific series of preferred stock under this prospectus, we will describe the terms of the preferred stock in the prospectus
supplement for such offering and will file a copy of the certificate establishing the terms of the preferred stock with the SEC. To the
extent required, this description will include:
| |
● |
the
title and stated value; |
| |
|
|
| |
● |
the
number of shares offered, the liquidation preference per share and the purchase price; |
| |
|
|
| |
● |
the
dividend rate(s), period(s) and/or payment date(s), or method(s) of calculation for such dividends; |
| |
|
|
| |
● |
whether
dividends will be cumulative or non-cumulative and, if cumulative, the date from which dividends will accumulate; |
| |
|
|
| |
● |
the
procedures for any auction and remarketing, if any; |
| |
|
|
| |
● |
the
provisions for a sinking fund, if any; |
| |
|
|
| |
● |
the
provisions for redemption, if applicable; |
| |
|
|
| |
● |
any
listing of the preferred stock on any securities exchange or market; |
| |
|
|
| |
● |
whether
the preferred stock will be convertible into our common stock, and, if applicable, the conversion price (or how it will be calculated)
and conversion period; |
| |
|
|
| |
● |
whether
the preferred stock will be exchangeable into debt securities, and, if applicable, the exchange price (or how it will be calculated)
and exchange period; |
| |
|
|
| |
● |
voting
rights, if any, of the preferred stock; |
| |
|
|
| |
● |
a
discussion of any material and/or special U.S. federal income tax considerations applicable to the preferred stock; |
| |
|
|
| |
● |
the
relative ranking and preferences of the preferred stock as to dividend rights and rights upon liquidation, dissolution or winding
up of our affairs; and |
| |
|
|
| |
● |
any
material limitations on issuance of any class or series of preferred stock ranking senior to or on a parity with the series of preferred
stock as to dividend rights and rights upon liquidation, dissolution or winding up of our affairs. |
Transfer
Agent and Registrar for Preferred Stock
The
transfer agent and registrar for any series or class of preferred stock will be set forth in each applicable prospectus supplement.
Anti-takeover
Effects of Delaware Law and our Certificate of Incorporation, as amended and Bylaws
Our
Certificate of Incorporation, as amended, and Bylaws, as amended contain provisions that could have the effect of discouraging potential
acquisition proposals or tender offers or delaying or preventing a change of control. These provisions are as follows:
| |
● |
they
provide that special meetings of stockholders may be called by the President, the board of directors or at the request by stockholders
of record owning at least thirty-three and one-third (33 1/3%) percent of the issued and outstanding voting shares of our Common
Stock; |
| |
● |
they
do not include a provision for cumulative voting in the election of directors. Under cumulative voting, a minority stockholder holding
a sufficient number of shares may be able to ensure the election of one or more directors. The absence of cumulative voting may have
the effect of limiting the ability of minority stockholders to effect changes in our board of directors; and |
| |
|
|
| |
● |
they
allow us to issue, without stockholder approval, up to 5,000,000 shares of preferred stock that could adversely affect the rights
and powers of the holders of our Common Stock. |
We
are subject to the provisions of Section 203 of the Delaware General Corporation Law, an anti-takeover law. Subject to certain exceptions,
the statute prohibits a publicly held Delaware corporation from engaging in a “business combination” with an “interested
stockholder” for a period of three years after the date of the transaction in which the person became an interested stockholder
unless:
| |
● |
prior
to such date, the board of directors of the corporation approved either the business combination or the transaction which resulted
in the stockholder becoming an interested stockholder; |
| |
|
|
| |
● |
upon
consummation of the transaction which resulted in the stockholder becoming an interested stockholder, the interested stockholder
owned at least eighty-five percent 85% of the voting stock of the corporation outstanding at the time the transaction commenced,
excluding for purposes of determining the number of shares outstanding those shares owned (1) by persons who are directors and also
officers and (2) by employee stock plans in which employee participants do not have the right to determine confidentially whether
shares held subject to the plan will be tendered in a tender or exchange offer; or |
| |
|
|
| |
● |
on
or after such date, the business combination is approved by the board of directors and authorized at an annual or special meeting
of stockholders, and not by written consent, by the affirmative vote of at least sixty-six and two-thirds percent 66 2/3% of the
outstanding voting stock that is not owned by the interested stockholder. |
Generally,
for purposes of Section 203, a “business combination” includes a merger, asset or stock sale, or other transaction resulting
in a financial benefit to the interested stockholder. An “interested stockholder” is a person who, together with affiliates
and associates, owns or, within three (3) years prior to the determination of interested stockholder status, owned fifteen percent (15%)
or more of a corporation’s outstanding voting securities.
Potential
Effects of Authorized but Unissued Stock
We
have shares of Common Stock and preferred stock available for future issuance without stockholder approval. We may utilize these additional
shares for a variety of corporate purposes, including future public offerings to raise additional capital, to facilitate corporate acquisitions
or payment as a dividend on the capital stock.
The
existence of unissued and unreserved Common Stock and preferred stock may enable our board of directors to issue shares to persons friendly
to current management or to issue preferred stock with terms that could render more difficult or discourage a third-party attempt to
obtain control of us by means of a merger, tender offer, proxy contest or otherwise, thereby protecting the continuity of our management.
In addition, the board of directors has the discretion to determine designations, rights, preferences, privileges and restrictions, including
voting rights, dividend rights, conversion rights, redemption privileges and liquidation preferences of each series of preferred stock,
all to the fullest extent permissible under the DGCL and subject to any limitations set forth in our Certificate of Incorporation. The
purpose of authorizing the board of directors to issue preferred stock and to determine the rights and preferences applicable to such
preferred stock is to eliminate delays associated with a stockholder vote on specific issuances. The issuance of preferred stock, while
providing desirable flexibility in connection with possible financings, acquisitions and other corporate purposes, could have the effect
of making it more difficult for a third-party to acquire, or could discourage a third-party from acquiring, a majority of our outstanding
voting stock.
Nasdaq
Listing
Our
common stock is traded on The Nasdaq Capital Market under the symbol “SONN.”
Exhibit
21.1
Subsidiaries
of Sonnet BioTherapeutics Holdings, Inc.
| Name: |
|
Jurisdiction
of Organization |
| Sonnet
BioTherapeutics, Inc. |
|
New
Jersey |
| Sonnet
BioTherapeutics CH SA |
|
Switzerland |
| SonnetBio
Pty Ltd |
|
Australia |
Exhibit
23.1
Consent
of Independent Registered Public Accounting Firm
We
consent to the incorporation by reference in the registration statements (Nos. 333-238542, 333-256183, 333-264832 and 333-274122) on
Form S-8, (Nos. 333-235301, 333-237795, 333-237354, 333-251406, 333-252049 and 333-267171) on Form S-3, and (Nos. 333-258092, 333-269307,
333-273516 and 333-274581) on Form S-1 of our report dated December 14, 2023, with respect to the consolidated financial statements of
Sonnet BioTherapeutics Holdings, Inc.
/s/
KPMG LLP
Philadelphia,
Pennsylvania
December
14, 2023
Exhibit
31.1
CERTIFICATION
OF PRINCIPAL EXECUTIVE OFFICER
PURSUANT
TO SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I,
Pankaj Mohan certify that:
1.
I have reviewed this annual report on Form 10-K of Sonnet BioTherapeutics Holdings, Inc. (the “Registrant”);
2.
Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary
to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the
period covered by this report;
3.
Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material
respects the financial condition, results of operations and cash flows of the Registrant as of, and for, the periods presented in this
report;
4.
The Registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures
(as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act
Rules 13a-15(f) and 15d-15(f)) for the Registrant and have:
a.
designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision,
to ensure that material information relating to the Registrant, including its consolidated subsidiaries, is made known to us by others
within those entities, particularly during the period in which this report is being prepared;
b.
designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under
our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements
for external purposes in accordance with generally accepted accounting principles;
c.
evaluated the effectiveness of the Registrant’s disclosure controls and procedures and presented in this report our conclusions
about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation;
and
d.
disclosed in this report any change in the Registrant’s internal control over financial reporting that occurred during the Registrant’s
most recent fiscal quarter (the Registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected,
or is reasonably likely to materially affect, the Registrant’s internal control over financial reporting; and
5.
The Registrant’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial
reporting, to the Registrant’s auditors and the audit committee of the Registrant’s board of directors (or persons performing
the equivalent functions):
a.
all significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are
reasonably likely to adversely affect the Registrant’s ability to record, process, summarize and report financial information;
and
b.
any fraud, whether or not material, that involves management or other employees who have a significant role in the Registrant’s
internal control over financial reporting.
| Date:
December 14, 2023 |
|
| |
/s/
Pankaj Mohan |
| |
Pankaj
Mohan |
| |
President
and Chief Executive Officer |
| |
(Principal
Executive Officer) |
Exhibit
31.2
CERTIFICATION
OF PRINCIPAL FINANCIAL OFFICER
PURSUANT
TO SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I,
Jay Cross certify that:
1.
I have reviewed this annual report on Form 10-K of Sonnet BioTherapeutics Holdings, Inc. (the “Registrant”);
2.
Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary
to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the
period covered by this report;
3.
Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material
respects the financial condition, results of operations and cash flows of the Registrant as of, and for, the periods presented in this
report;
4.
The Registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures
(as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act
Rules 13a-15(f) and 15d-15(f)) for the Registrant and have:
a.
designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision,
to ensure that material information relating to the Registrant, including its consolidated subsidiaries, is made known to us by others
within those entities, particularly during the period in which this report is being prepared;
b.
designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under
our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements
for external purposes in accordance with generally accepted accounting principles;
c.
evaluated the effectiveness of the Registrant’s disclosure controls and procedures and presented in this report our conclusions
about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation;
and
d.
disclosed in this report any change in the Registrant’s internal control over financial reporting that occurred during the Registrant’s
most recent fiscal quarter (the Registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected,
or is reasonably likely to materially affect, the Registrant’s internal control over financial reporting; and
5.
The Registrant’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial
reporting, to the Registrant’s auditors and the audit committee of the Registrant’s board of directors (or persons performing
the equivalent functions):
a.
all significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are
reasonably likely to adversely affect the Registrant’s ability to record, process, summarize and report financial information;
and
b.
any fraud, whether or not material, that involves management or other employees who have a significant role in the Registrant’s
internal control over financial reporting.
| Date:
December 14, 2023 |
|
| |
/s/
Jay Cross |
| |
Jay
Cross |
| |
Chief
Financial Officer |
| |
(Principal
Financial and Accounting Officer) |
Exhibit
32.1
CERTIFICATION
PURSUANT TO 18 U.S.C. SECTION 1350,
AS
ADOPTED PURSUANT TO
SECTION
906 OF THE SARBANES-OXLEY ACT OF 2002
In
connection with the annual report of Sonnet BioTherapeutics Holdings, Inc. (the “Company”) on Form 10-K for the year ended
September 30, 2023 as filed with the Securities and Exchange Commission on the date hereof (the “Report”), I, Pankaj Mohan,
President and Chief Executive Officer of the Company, certify, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906
of the Sarbanes-Oxley Act of 2002, that, to my knowledge:
| |
(1) | The Report fully
complies with the requirements of Section 13(a) or 15(d), as applicable, of the Securities Exchange Act of 1934, as amended; and |
| |
(2) | The information
contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company. |
Date:
December 14, 2023
| /s/
Pankaj Mohan |
|
| Pankaj
Mohan |
|
| President
and Chief Executive Officer |
|
(Principal
Executive Officer)
|
|
The
foregoing certification is being furnished solely pursuant to 18 U.S.C. Section 1350 and is not being filed as part of the Report or
as a separate disclosure document.
Exhibit
32.2
CERTIFICATION
PURSUANT TO 18 U.S.C. SECTION 1350,
AS
ADOPTED PURSUANT TO
SECTION
906 OF THE SARBANES-OXLEY ACT OF 2002
In
connection with the annual report of Sonnet BioTherapeutics Holdings, Inc. (the “Company”) on Form 10-K for the year ended
September 30, 2023 as filed with the Securities and Exchange Commission on the date hereof (the “Report”), I, Jay Cross,
Chief Financial Officer of the Company, certify, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley
Act of 2002, that, to my knowledge:
| |
(1) | The Report fully
complies with the requirements of Section 13(a) or 15(d), as applicable, of the Securities Exchange Act of 1934, as amended; and |
| |
(2) | The information
contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company. |
Date:
December 14, 2023
| /s/
Jay Cross |
|
| Jay
Cross |
|
| Chief
Financial Officer |
|
| (Principal
Financial and Accounting Officer) |
|
The
foregoing certification is being furnished solely pursuant to 18 U.S.C. Section 1350 and is not being filed as part of the Report or
as a separate disclosure document.
Exhibit
97.1
SONNET
BIOTHERAPEUTICS, INC.
COMPENSATION
RECOVERY POLICY
(Adopted
and approved on November 21, 2023)
1.
Purpose
Sonnet
BioTherapeutics, Inc. (collectively with its subsidiaries, the “Company”) is committed to promoting high standards
of honest and ethical business conduct and compliance with applicable laws, rules and regulations. As part of this commitment, the Company
has adopted this Compensation Recovery Policy (this “Policy”). This Policy is designed to comply with the requirements
of Section 10D of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), Rule 10D-1 promulgated thereunder
and the rules of the national securities exchange on which the Company’s securities are traded and explains when the Company will
pursue recovery of Incentive Compensation awarded or paid to a Covered Person. Please refer to Exhibit A attached hereto (the “Definitions
Exhibit”) for the definitions of capitalized terms used throughout this Policy.
2.
Recovery of Recoverable Incentive Compensation
In
the event of a Restatement, the Company will pursue, reasonably promptly, recovery of all Recoverable Incentive Compensation from a Covered
Person without regard to such Covered Person’s individual knowledge or responsibility related to the Restatement. Notwithstanding
the foregoing, if the Company is otherwise required by this Policy to undertake a Restatement, the Company will not be required to recover
the Recoverable Incentive Compensation if the Compensation Committee determines, after exercising a normal due process review of all
the relevant facts and circumstances, that (a) a Recovery Exception exists and (b) it would be impracticable to seek such recovery under
such facts and circumstances.
If
such Recoverable Incentive Compensation was not awarded or paid on a formulaic basis, the Company will pursue recovery of the amount
that the Compensation Committee determines in good faith should be recovered.
3.
Other Actions
The
Compensation Committee may, subject to applicable law, pursue recovery of Recoverable Incentive Compensation in the manner it chooses,
including by pursuing reimbursement from the Covered Person of all or part of the compensation awarded or paid, by electing to withhold
unpaid compensation, by set-off, or by rescinding or canceling unvested stock or option awards.
In
the reasonable exercise of its business judgment under this Policy, the Compensation Committee may in its sole discretion determine whether
and to what extent additional action is appropriate to address the circumstances surrounding a Restatement to minimize the likelihood
of any recurrence and to impose such other discipline as it deems appropriate.
4.
No Indemnification or Reimbursement
As
required by applicable law, notwithstanding the terms of any other policy, program, agreement or arrangement, in no event will the Company
or any of its affiliates indemnify or reimburse a Covered Person for any loss of Recoverable Incentive Compensation under this Policy
and, to the extent prohibited by law, neither the Company nor any of its affiliates will pay premiums on any insurance policy that would
cover a Covered Person’s potential obligations with respect to Recoverable Incentive Compensation under this Policy.
5.
Administration of Policy
The
Compensation Committee will have full authority to administer this Policy. The Compensation Committee will, subject to the provisions
of this Policy and Rule 10D-1 of the Exchange Act, and the Company’s applicable exchange listing standards, make such determinations
and interpretations and take such actions in connection with this Policy as it deems necessary, appropriate or advisable. It is intended
that this Policy be interpreted in a manner that is consistent with the requirements of Section 10D of the Exchange Act, Rule 10D-1 thereunder
and any applicable rules or standards adopted by the Securities and Exchange Commission or any national securities exchange on which
the Company’s securities are listed. All determinations and interpretations made by the Compensation Committee will be final, binding
and conclusive.
6.
Other Claims and Rights
The
requirements of this Policy are in addition to, and not in lieu of, any legal and equitable claims the Company or any of its affiliates
may have or any actions that may be imposed by law enforcement agencies, regulators, administrative bodies, or other authorities. Further,
the exercise by the Compensation Committee of any rights pursuant to this Policy will not impact any other rights that the Company or
any of its affiliates may have with respect to any Covered Person subject to this Policy.
7.
Acknowledgement by Covered Persons; Condition to Eligibility for Incentive Compensation
The
Company will provide notice and seek acknowledgement of this Policy from each Covered Person, provided that the failure to provide such
notice or obtain such acknowledgement will have no impact on the applicability or enforceability of this Policy. After the Effective
Date (and also with respect to any Incentive Compensation Received on or after October 2, 2023 pursuant to a preexisting contract or
arrangement), any grant of Incentive Compensation to a Covered Person will be deemed to have been made subject to the terms of this Policy,
whether or not such Policy is specifically referenced in the documentation relating to such grant and this Policy shall be deemed to
constitute an integral part of the terms of any such grant. All Incentive Compensation subject to this Policy will remain subject to
this policy, even if already paid, until the Policy ceases to apply to such Incentive Compensation and any other vesting conditions applicable
to such Incentive Compensation are satisfied.
8.
Amendment; Termination
The
Board or the Compensation Committee may amend or terminate this Policy at any time. In the event that Section 10D of the Exchange Act,
Rule 10D-1 thereunder or the rules of the national securities exchange on which the Company’s securities are traded are modified
or supplemented, whether by law, regulation or legal interpretation, such modification or supplement shall be deemed to modify or supplement
this Policy to the maximum extent permitted by applicable law.
9.
Effectiveness
Except
as otherwise determined in writing by the Compensation Committee, this Policy will apply to any Incentive Compensation that is Received
by a Covered Person on or after the Effective Date. This Policy will survive and continue notwithstanding any termination of a Covered
Person’s employment with the Company and its affiliates.
10.
Successors
This
Policy shall be binding and enforceable against all Covered Persons and their successors, beneficiaries, heirs, executors, administrators,
or other legal representatives.
Exhibit
A
SONNET
BIOTHERAPEUTICS, INC.
COMPENSATION
RECOVERY POLICY
DEFINITIONS
EXHIBIT
“Applicable
Period” means the three completed fiscal years of the Company immediately preceding the earlier of (i) the date the Board,
a committee of the Board, or the officer or officers of the Company authorized to take such action if Board action is not required, concludes
(or reasonably should have concluded) that a Restatement is required or (ii) the date a court, regulator, or other legally authorized
body directs the Company to prepare a Restatement. The “Applicable Period” also includes any transition period (that results
from a change in the Company’s fiscal year) within or immediately following the three completed fiscal years identified in the
preceding sentence.
“Board”
means the Board of Directors of the Company.
“Compensation
Committee” means the Company’s committee of independent directors responsible for executive compensation decisions, or
in the absence of such a committee, a majority of the independent directors serving on the Board.
“Covered
Person” means any person who is, or was at any time, during the Applicable Period, an Executive Officer of the Company. For
the avoidance of doubt, a Covered Person may include a former Executive Officer that left the Company, retired, or transitioned to an
employee role (including after serving as an Executive Officer in an interim capacity) during the Applicable Period.
“Effective
Date” means December 1, 2023.
“Executive
Officer” means the Company’s president, principal executive officer, principal financial officer, principal accounting
officer (or if there is no such accounting officer, the controller), any vice-president in charge of a principal business unit, division,
or function (such as sales, administration, or finance), any other officer who performs a policy-making function, or any other person
(including an officer of the Company’s parent(s) or subsidiaries) who performs similar policy-making functions for the Company.
“Financial
Reporting Measure” means a measure that is determined and presented in accordance with the accounting principles used in preparing
the Company’s financial statements, and any measure that is derived wholly or in part from such measure (including but not limited
to, “non-GAAP” financial measures, such as those appearing in the Company’s earnings releases or Management Discussion
and Analysis). Stock price and total shareholder return (and any measures derived wholly or in part therefrom) shall be considered Financial
Reporting Measures.
“Recovery
Exception:” A recovery of Recoverable Incentive Compensation shall be subject to a “Recovery Exception” if the
Compensation Committee determines in good faith that: (i) pursuing such recovery would violate home country law of the jurisdiction of
incorporation of the Company where that law was adopted prior to November 28, 2022 and the Company provides an opinion of home country
counsel to that effect acceptable to the Company’s applicable listing exchange; (ii) the direct expense paid to a third party to
assist in enforcing this Policy would exceed the Recoverable Incentive Compensation and the Company has (A) made a reasonable attempt
to recover such amounts and (B) provided documentation of such attempts to recover to the Company’s applicable listing exchange;
or (iii) recovery would likely cause an otherwise tax-qualified retirement plan, under which benefits are broadly available to employees
of the Company, to fail to meet the requirements of Section 401(a)(13) or Section 411(a) of the Internal Revenue Code of 1986, as amended,
and regulations thereunder.
“Incentive
Compensation” means any compensation that is granted, earned, or vested based wholly or in part upon the attainment of a Financial
Reporting Measure. Incentive Compensation does not include any base salaries (except with respect to any salary increases earned wholly
or in part based on the attainment of a Financial Reporting Measure performance goal); bonuses paid solely at the discretion of the Compensation
Committee or Board that are not paid from a “bonus pool” that is determined by satisfying a Financial Reporting Measure performance
goal; bonuses paid solely upon satisfying one or more subjective standards and/or completion of a specified employment period; non-equity
incentive plan awards earned solely upon satisfying one or more strategic measures or operational measures; and equity awards that vest
solely based on the passage of time and/or attaining one or more non-Financial Reporting Measures. Incentive Compensation includes any
Incentive Compensation Received on or after October 2, 2023 pursuant to a preexisting contract or arrangement.
“Received:”
Incentive Compensation is deemed “Received” in the Company’s fiscal period during which the Financial Reporting Measure
specified in the Incentive Compensation award is attained, even if the payment or grant of the Incentive Compensation occurs after the
end of that period.
“Recoverable
Incentive Compensation” means the amount of any Incentive Compensation (calculated on a pre-tax basis) Received by a Covered
Person during the Applicable Period that is in excess of the amount that otherwise would have been Received if the calculation were based
on the Restatement. For Incentive Compensation based on (or derived from) stock price or total shareholder return where the amount of
Recoverable Incentive Compensation is not subject to mathematical recalculation directly from the information in the applicable Restatement,
the amount will be determined by the Compensation Committee based on a reasonable estimate of the effect of the Restatement on the stock
price or total shareholder return upon which the Incentive Compensation was Received (in which case, the Company will maintain documentation
of such determination of that reasonable estimate and provide such documentation to the Company’s applicable listing exchange).
“Restatement”
means an accounting restatement of any of the Company’s financial statements filed with the Securities and Exchange Commission
under the Exchange Act, or the Securities Act of 1933, as amended, due to the Company’s material noncompliance with any financial
reporting requirement under U.S. securities laws, regardless of whether the Company or Covered Person misconduct was the cause for such
restatement. “Restatement” includes any required accounting restatement to correct an error in previously issued financial
statements that is material to the previously issued financial statements (commonly referred to as “Big R” restatements),
or that would result in a material misstatement if the error were corrected in the current period or left uncorrected in the current
period (commonly referred to as “little r” restatements).
v3.23.3
Cover - USD ($)
$ in Millions |
12 Months Ended |
|
|
Sep. 30, 2023 |
Dec. 11, 2023 |
Mar. 31, 2023 |
| Cover [Abstract] |
|
|
|
| Document Type |
10-K
|
|
|
| Amendment Flag |
false
|
|
|
| Document Annual Report |
true
|
|
|
| Document Transition Report |
false
|
|
|
| Document Period End Date |
Sep. 30, 2023
|
|
|
| Document Fiscal Period Focus |
FY
|
|
|
| Document Fiscal Year Focus |
2023
|
|
|
| Current Fiscal Year End Date |
--09-30
|
|
|
| Entity File Number |
001-35570
|
|
|
| Entity Registrant Name |
SONNET
BIOTHERAPEUTICS HOLDINGS, INC.
|
|
|
| Entity Central Index Key |
0001106838
|
|
|
| Entity Tax Identification Number |
20-2932652
|
|
|
| Entity Incorporation, State or Country Code |
DE
|
|
|
| Entity Address, Address Line One |
100
Overlook Center
|
|
|
| Entity Address, Address Line Two |
Suite 102
|
|
|
| Entity Address, City or Town |
Princeton
|
|
|
| Entity Address, State or Province |
NJ
|
|
|
| Entity Address, Postal Zip Code |
08540
|
|
|
| City Area Code |
(609)
|
|
|
| Local Phone Number |
375-2227
|
|
|
| Title of 12(b) Security |
Common Stock, $0.0001
par value per share
|
|
|
| Trading Symbol |
SONN
|
|
|
| Security Exchange Name |
NASDAQ
|
|
|
| Entity Well-known Seasoned Issuer |
No
|
|
|
| Entity Voluntary Filers |
No
|
|
|
| Entity Current Reporting Status |
Yes
|
|
|
| Entity Interactive Data Current |
Yes
|
|
|
| Entity Filer Category |
Non-accelerated Filer
|
|
|
| Entity Small Business |
true
|
|
|
| Entity Emerging Growth Company |
false
|
|
|
| Entity Shell Company |
false
|
|
|
| Entity Public Float |
|
|
$ 7.3
|
| Entity Common Stock, Shares Outstanding |
|
3,069,516
|
|
| Documents Incorporated by Reference [Text Block] |
None
|
|
|
| ICFR Auditor Attestation Flag |
false
|
|
|
| Document Financial Statement Error Correction [Flag] |
false
|
|
|
| Auditor Firm ID |
185
|
|
|
| Auditor Name |
KPMG LLP
|
|
|
| Auditor Location |
Philadelphia, Pennsylvania
|
|
|
| X |
- DefinitionBoolean flag that is true when the XBRL content amends previously-filed or accepted submission.
| Name: |
dei_AmendmentFlag |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionPCAOB issued Audit Firm Identifier Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 10-K
-Number 249
-Section 310
Reference 2: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 20-F
-Number 249
-Section 220
-Subsection f
Reference 3: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 40-F
-Number 249
-Section 240
-Subsection f
| Name: |
dei_AuditorFirmId |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:nonemptySequenceNumberItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- ReferencesReference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 10-K
-Number 249
-Section 310
Reference 2: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 20-F
-Number 249
-Section 220
-Subsection f
Reference 3: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 40-F
-Number 249
-Section 240
-Subsection f
| Name: |
dei_AuditorLocation |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:internationalNameItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- ReferencesReference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 10-K
-Number 249
-Section 310
Reference 2: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 20-F
-Number 249
-Section 220
-Subsection f
Reference 3: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 40-F
-Number 249
-Section 240
-Subsection f
| Name: |
dei_AuditorName |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:internationalNameItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionEnd date of current fiscal year in the format --MM-DD.
| Name: |
dei_CurrentFiscalYearEndDate |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:gMonthDayItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionBoolean flag that is true only for a form used as an annual report. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 10-K
-Number 249
-Section 310
Reference 2: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 20-F
-Number 249
-Section 220
-Subsection f
Reference 3: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 40-F
-Number 249
-Section 240
-Subsection f
| Name: |
dei_DocumentAnnualReport |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionIndicates whether any of the financial statement period in the filing include a restatement due to error correction. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Regulation S-K
-Number 229
-Section 402
-Subsection w
Reference 2: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 10-K
-Number 249
-Section 310
Reference 3: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 20-F
-Number 249
-Section 220
-Subsection f
Reference 4: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 40-F
-Number 249
-Section 240
-Subsection f
| Name: |
dei_DocumentFinStmtErrorCorrectionFlag |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionFiscal period values are FY, Q1, Q2, and Q3. 1st, 2nd and 3rd quarter 10-Q or 10-QT statements have value Q1, Q2, and Q3 respectively, with 10-K, 10-KT or other fiscal year statements having FY.
| Name: |
dei_DocumentFiscalPeriodFocus |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:fiscalPeriodItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThis is focus fiscal year of the document report in YYYY format. For a 2006 annual report, which may also provide financial information from prior periods, fiscal 2006 should be given as the fiscal year focus. Example: 2006.
| Name: |
dei_DocumentFiscalYearFocus |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:gYearItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionFor the EDGAR submission types of Form 8-K: the date of the report, the date of the earliest event reported; for the EDGAR submission types of Form N-1A: the filing date; for all other submission types: the end of the reporting or transition period. The format of the date is YYYY-MM-DD.
| Name: |
dei_DocumentPeriodEndDate |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:dateItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionBoolean flag that is true only for a form used as a transition report. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Forms 10-K, 10-Q, 20-F
-Number 240
-Section 13
-Subsection a-1
| Name: |
dei_DocumentTransitionReport |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe type of document being provided (such as 10-K, 10-Q, 485BPOS, etc). The document type is limited to the same value as the supporting SEC submission type, or the word 'Other'.
| Name: |
dei_DocumentType |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:submissionTypeItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDocuments incorporated by reference. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-23
| Name: |
dei_DocumentsIncorporatedByReferenceTextBlock |
| Namespace Prefix: |
dei_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAddress Line 1 such as Attn, Building Name, Street Name
| Name: |
dei_EntityAddressAddressLine1 |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:normalizedStringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAddress Line 2 such as Street or Suite number
| Name: |
dei_EntityAddressAddressLine2 |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:normalizedStringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Definition
+ References
+ Details
| Name: |
dei_EntityAddressCityOrTown |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:normalizedStringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionCode for the postal or zip code
| Name: |
dei_EntityAddressPostalZipCode |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:normalizedStringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionName of the state or province.
| Name: |
dei_EntityAddressStateOrProvince |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:stateOrProvinceItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionA unique 10-digit SEC-issued value to identify entities that have filed disclosures with the SEC. It is commonly abbreviated as CIK. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-2
| Name: |
dei_EntityCentralIndexKey |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:centralIndexKeyItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionIndicate number of shares or other units outstanding of each of registrant's classes of capital or common stock or other ownership interests, if and as stated on cover of related periodic report. Where multiple classes or units exist define each class/interest by adding class of stock items such as Common Class A [Member], Common Class B [Member] or Partnership Interest [Member] onto the Instrument [Domain] of the Entity Listings, Instrument.
| Name: |
dei_EntityCommonStockSharesOutstanding |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionIndicate 'Yes' or 'No' whether registrants (1) have filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that registrants were required to file such reports), and (2) have been subject to such filing requirements for the past 90 days. This information should be based on the registrant's current or most recent filing containing the related disclosure.
| Name: |
dei_EntityCurrentReportingStatus |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:yesNoItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionIndicate if registrant meets the emerging growth company criteria. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-2
| Name: |
dei_EntityEmergingGrowthCompany |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionCommission file number. The field allows up to 17 characters. The prefix may contain 1-3 digits, the sequence number may contain 1-8 digits, the optional suffix may contain 1-4 characters, and the fields are separated with a hyphen.
| Name: |
dei_EntityFileNumber |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:fileNumberItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionIndicate whether the registrant is one of the following: Large Accelerated Filer, Accelerated Filer, Non-accelerated Filer. Definitions of these categories are stated in Rule 12b-2 of the Exchange Act. This information should be based on the registrant's current or most recent filing containing the related disclosure. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-2
| Name: |
dei_EntityFilerCategory |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:filerCategoryItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTwo-character EDGAR code representing the state or country of incorporation.
| Name: |
dei_EntityIncorporationStateCountryCode |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:edgarStateCountryItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionBoolean flag that is true when the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Regulation S-T
-Number 232
-Section 405
| Name: |
dei_EntityInteractiveDataCurrent |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:yesNoItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe aggregate market value of the voting and non-voting common equity held by non-affiliates computed by reference to the price at which the common equity was last sold, or the average bid and asked price of such common equity, as of the last business day of the registrant's most recently completed second fiscal quarter.
| Name: |
dei_EntityPublicFloat |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionThe exact name of the entity filing the report as specified in its charter, which is required by forms filed with the SEC. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-2
| Name: |
dei_EntityRegistrantName |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:normalizedStringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionBoolean flag that is true when the registrant is a shell company as defined in Rule 12b-2 of the Exchange Act. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-2
| Name: |
dei_EntityShellCompany |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionIndicates that the company is a Smaller Reporting Company (SRC). Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-2
| Name: |
dei_EntitySmallBusiness |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe Tax Identification Number (TIN), also known as an Employer Identification Number (EIN), is a unique 9-digit value assigned by the IRS. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-2
| Name: |
dei_EntityTaxIdentificationNumber |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:employerIdItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionIndicate 'Yes' or 'No' if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
| Name: |
dei_EntityVoluntaryFilers |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:yesNoItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionIndicate 'Yes' or 'No' if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Is used on Form Type: 10-K, 10-Q, 8-K, 20-F, 6-K, 10-K/A, 10-Q/A, 20-F/A, 6-K/A, N-CSR, N-Q, N-1A. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Securities Act
-Number 230
-Section 405
| Name: |
dei_EntityWellKnownSeasonedIssuer |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:yesNoItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- ReferencesReference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 10-K
-Number 249
-Section 310
Reference 2: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 20-F
-Number 249
-Section 220
-Subsection f
Reference 3: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Form 40-F
-Number 249
-Section 240
-Subsection f
| Name: |
dei_IcfrAuditorAttestationFlag |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionLocal phone number for entity.
| Name: |
dei_LocalPhoneNumber |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:normalizedStringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTitle of a 12(b) registered security. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b
| Name: |
dei_Security12bTitle |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:securityTitleItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionName of the Exchange on which a security is registered. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection d1-1
| Name: |
dei_SecurityExchangeName |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:edgarExchangeCodeItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTrading symbol of an instrument as listed on an exchange.
| Name: |
dei_TradingSymbol |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:tradingSymbolItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Consolidated Balance Sheets - USD ($)
|
Sep. 30, 2023 |
Sep. 30, 2022 |
| Current assets: |
|
|
| Cash and cash equivalents |
$ 2,274,259
|
$ 3,052,879
|
| Prepaid expenses and other current assets |
1,677,396
|
1,643,743
|
| Incentive tax receivable |
786,574
|
717,305
|
| Total current assets |
4,738,229
|
5,413,927
|
| Property and equipment, net |
33,366
|
46,211
|
| Operating lease right-of-use asset |
193,689
|
256,594
|
| Deferred offering costs |
49,988
|
113,280
|
| Other assets |
414,206
|
|
| Total assets |
5,429,478
|
5,830,012
|
| Current liabilities: |
|
|
| Accounts payable |
2,201,999
|
4,752,340
|
| Accrued expenses and other current liabilities |
3,230,922
|
3,193,972
|
| Current portion of operating lease liability |
73,048
|
51,328
|
| Deferred income |
18,626
|
166,431
|
| Total current liabilities |
5,524,595
|
8,164,819
|
| Operating lease liability, net of current portion |
130,863
|
203,912
|
| Total liabilities |
5,655,458
|
8,368,731
|
| Commitments and contingencies (Note 4) |
|
|
| Stockholders’ deficit: |
|
|
| Common stock, $0.0001 par value: 125,000,000 shares authorized; 1,750,426 and 251,955 issued and outstanding at September 30, 2023 and 2022, respectively |
175
|
25
|
| Additional paid-in capital |
110,017,598
|
88,872,315
|
| Accumulated deficit |
(110,243,753)
|
(91,411,059)
|
| Total stockholders’ deficit |
(225,980)
|
(2,538,719)
|
| Total liabilities and stockholders’ deficit |
5,429,478
|
5,830,012
|
| Related Party [Member] |
|
|
| Current liabilities: |
|
|
| Related party notes |
|
$ 748
|
| X |
- DefinitionIncentive tax receivable current.
| Name: |
SONN_IncentiveTaxReceivableCurrent |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of liabilities incurred to vendors for goods and services received, and accrued liabilities classified as other, payable within one year or the normal operating cycle, if longer.
| Name: |
us-gaap_AccountsPayableAndOtherAccruedLiabilitiesCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionCarrying value as of the balance sheet date of liabilities incurred (and for which invoices have typically been received) and payable to vendors for goods and services received that are used in an entity's business. Used to reflect the current portion of the liabilities (due within one year or within the normal operating cycle if longer). Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.19(a))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_AccountsPayableCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of excess of issue price over par or stated value of stock and from other transaction involving stock or stockholder. Includes, but is not limited to, additional paid-in capital (APIC) for common and preferred stock. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 2: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(18))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(30)(a)(1))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_AdditionalPaidInCapital |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionSum of the carrying amounts as of the balance sheet date of all assets that are recognized. Assets are probable future economic benefits obtained or controlled by an entity as a result of past transactions or events. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (bb)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481203/810-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 25
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481231/810-10-45-25
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 6: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 7: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 12
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-12
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(12))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(8))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(18))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 13: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 14: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 20: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(B))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 21: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 22: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 23: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481404/852-10-50-7
Reference 24: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 25: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 26: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 942
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-03(11))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479853/942-210-S99-1
| Name: |
us-gaap_Assets |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionSum of the carrying amounts as of the balance sheet date of all assets that are expected to be realized in cash, sold, or consumed within one year (or the normal operating cycle, if longer). Assets are probable future economic benefits obtained or controlled by an entity as a result of past transactions or events. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (bb)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481203/810-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 25
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481231/810-10-45-25
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 6: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483467/210-10-45-1
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(9))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 10: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 11: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(B))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 20: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481404/852-10-50-7
| Name: |
us-gaap_AssetsCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- References
+ Details
| Name: |
us-gaap_AssetsCurrentAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of short-term, highly liquid investments that are both readily convertible to known amounts of cash and so near their maturity that they present insignificant risk of changes in value because of changes in interest rates. Excludes cash and cash equivalents within disposal group and discontinued operation. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(2))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 2: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(1))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_CashEquivalentsAtCarryingValue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionRepresents the caption on the face of the balance sheet to indicate that the entity has entered into (1) purchase or supply arrangements that will require expending a portion of its resources to meet the terms thereof, and (2) is exposed to potential losses or, less frequently, gains, arising from (a) possible claims against a company's resources due to future performance under contract terms, and (b) possible losses or likely gains from uncertainties that will ultimately be resolved when one or more future events that are deemed likely to occur do occur or fail to occur. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(19))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(15))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 942
-SubTopic 210
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-03.17)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479853/942-210-S99-1
Reference 4: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.25)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_CommitmentsAndContingencies |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAggregate par or stated value of issued nonredeemable common stock (or common stock redeemable solely at the option of the issuer). This item includes treasury stock repurchased by the entity. Note: elements for number of nonredeemable common shares, par value and other disclosure concepts are in another section within stockholders' equity. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(22))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_CommonStockValue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of deferred cost, excluding capitalized cost related to contract with customer; classified as noncurrent. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(17))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 942
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-03(10))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479853/942-210-S99-1
| Name: |
us-gaap_DeferredCosts |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of deferred income excluding obligation to transfer product and service to customer for which consideration has been received or is receivable, classified as current. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Name Accounting Standards Codification
-Section 25
-Paragraph 2
-SubTopic 10
-Topic 470
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481174/470-10-25-2
| Name: |
us-gaap_DeferredIncomeCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionSum of the carrying amounts as of the balance sheet date of all liabilities that are recognized. Liabilities are probable future sacrifices of economic benefits arising from present obligations of an entity to transfer assets or provide services to other entities in the future. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481203/810-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 25
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481231/810-10-45-25
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (bb)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481203/810-10-50-3
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 7: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 12
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-12
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(14))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 10: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(B))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 19: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481404/852-10-50-7
Reference 20: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481404/852-10-50-7
Reference 21: http://www.xbrl.org/2003/role/exampleRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 22: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.19-26)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_Liabilities |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of liabilities and equity items, including the portion of equity attributable to noncontrolling interests, if any. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(25))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 3: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 4: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 5: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 6: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 942
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-03(23))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479853/942-210-S99-1
Reference 7: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(32))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_LiabilitiesAndStockholdersEquity |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionTotal obligations incurred as part of normal operations that are expected to be paid during the following twelve months or within one business cycle, if longer. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481203/810-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 25
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481231/810-10-45-25
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 810
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (bb)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481203/810-10-50-3
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 7: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 5
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483467/210-10-45-5
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 10: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(B))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 19: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481404/852-10-50-7
Reference 20: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481404/852-10-50-7
Reference 21: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.21)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_LiabilitiesCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- References
+ Details
| Name: |
us-gaap_LiabilitiesCurrentAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionSum of the carrying values as of the balance sheet date of the portions of long-term notes payable due within one year or the operating cycle if longer. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.19,20)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_NotesPayableCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionPresent value of lessee's discounted obligation for lease payments from operating lease, classified as current. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479041/842-20-45-1
| Name: |
us-gaap_OperatingLeaseLiabilityCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionPresent value of lessee's discounted obligation for lease payments from operating lease, classified as noncurrent. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479041/842-20-45-1
| Name: |
us-gaap_OperatingLeaseLiabilityNoncurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of lessee's right to use underlying asset under operating lease. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479041/842-20-45-1
| Name: |
us-gaap_OperatingLeaseRightOfUseAsset |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of noncurrent assets classified as other. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(17))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_OtherAssetsNoncurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of asset related to consideration paid in advance for costs that provide economic benefits in future periods, and amount of other assets that are expected to be realized or consumed within one year or the normal operating cycle, if longer. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(9))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_PrepaidExpenseAndOtherAssetsCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount after accumulated depreciation, depletion and amortization of physical assets used in the normal conduct of business to produce goods and services and not intended for resale. Examples include, but are not limited to, land, buildings, machinery and equipment, office equipment, and furniture and fixtures. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-SubTopic 10
-Topic 360
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482099/360-10-50-1
Reference 2: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(8))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 942
-SubTopic 360
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480842/942-360-50-1
| Name: |
us-gaap_PropertyPlantAndEquipmentNet |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of accumulated undistributed earnings (deficit). Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 2
-Subparagraph (g)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480016/944-40-65-2
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 2
-Subparagraph (h)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480016/944-40-65-2
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480990/946-20-50-11
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(23)(a)(4))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(17))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 7: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 8: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(30)(a)(3))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_RetainedEarningsAccumulatedDeficit |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of equity (deficit) attributable to parent. Excludes temporary equity and equity attributable to noncontrolling interest. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 2: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 12
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-12
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(19))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.6-05(4))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-2
Reference 5: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(4)(b))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(6))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(7))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 8: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 9: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 10: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 11: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 12: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(31))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 13: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(30))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 14: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 310
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SAB Topic 4.E)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480418/310-10-S99-2
| Name: |
us-gaap_StockholdersEquity |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- References
+ Details
| Name: |
us-gaap_StockholdersEquityIncludingPortionAttributableToNoncontrollingInterestAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Consolidated Balance Sheets (Parenthetical) - $ / shares
|
Sep. 30, 2023 |
Sep. 30, 2022 |
| Statement of Financial Position [Abstract] |
|
|
| Common stock, par value |
$ 0.0001
|
$ 0.0001
|
| Common stock, shares authorized |
125,000,000
|
125,000,000
|
| Common stock, shares issued |
1,750,426
|
251,955
|
| Common stock, shares outstanding |
1,750,426
|
251,955
|
| X |
- DefinitionFace amount or stated value per share of common stock. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_CommonStockParOrStatedValuePerShare |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionThe maximum number of common shares permitted to be issued by an entity's charter and bylaws. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(16)(a))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_CommonStockSharesAuthorized |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionTotal number of common shares of an entity that have been sold or granted to shareholders (includes common shares that were issued, repurchased and remain in the treasury). These shares represent capital invested by the firm's shareholders and owners, and may be all or only a portion of the number of shares authorized. Shares issued include shares outstanding and shares held in the treasury. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_CommonStockSharesIssued |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionNumber of shares of common stock outstanding. Common stock represent the ownership interest in a corporation. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.6-05(4))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-2
Reference 3: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(4)(b))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(16)(a))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 5: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(7))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 6: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_CommonStockSharesOutstanding |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- References
+ Details
| Name: |
us-gaap_StatementOfFinancialPositionAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Consolidated Statements of Operations - USD ($)
|
12 Months Ended |
Sep. 30, 2023 |
Sep. 30, 2022 |
| Income Statement [Abstract] |
|
|
| Collaboration revenue |
$ 147,805
|
$ 349,943
|
| Operating expenses: |
|
|
| Research and development |
11,814,690
|
21,444,019
|
| General and administrative |
7,125,732
|
8,575,283
|
| Total operating expense |
18,940,422
|
30,019,302
|
| Loss from operations |
(18,792,617)
|
(29,669,359)
|
| Foreign exchange loss |
(40,077)
|
(52,482)
|
| Net loss |
$ (18,832,694)
|
$ (29,721,841)
|
| Per share information: |
|
|
| Net loss per share, basic |
$ (18.14)
|
$ (150.52)
|
| Net loss per share, diluted |
$ (18.14)
|
$ (150.52)
|
| Weighted average shares outstanding, basic |
1,038,188
|
197,462
|
| Weighted average shares outstanding, diluted |
1,038,188
|
197,462
|
| X |
- References
+ Details
| Name: |
us-gaap_EarningsPerShareAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe amount of net income (loss) for the period per each share of common stock or unit outstanding during the reporting period. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 15
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482635/260-10-55-15
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (e)(4)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-7
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-2
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 60B
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-60B
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-4
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-10
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.5-03(25))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-2
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 942
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-04(27))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483589/942-220-S99-1
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-04(23))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483586/944-220-S99-1
Reference 16: http://www.xbrl.org/2003/role/exampleRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 52
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482635/260-10-55-52
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-7
| Name: |
us-gaap_EarningsPerShareBasic |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe amount of net income (loss) for the period available to each share of common stock or common unit outstanding during the reporting period and to each share or unit that would have been outstanding assuming the issuance of common shares or units for all dilutive potential common shares or units outstanding during the reporting period. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 15
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482635/260-10-55-15
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (e)(4)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-7
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-2
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 60B
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-60B
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-4
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.5-03(25))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-2
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 942
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-04(27))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483589/942-220-S99-1
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-04(23))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483586/944-220-S99-1
Reference 15: http://www.xbrl.org/2003/role/exampleRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 52
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482635/260-10-55-52
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-7
| Name: |
us-gaap_EarningsPerShareDiluted |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount, before tax, of realized and unrealized gain (loss) from foreign currency transaction. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-07(7))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 20
-Name Accounting Standards Codification
-Section 35
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482014/830-20-35-1
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 20
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481956/830-20-45-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481926/830-20-50-1
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 17
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481839/830-10-45-17
| Name: |
us-gaap_ForeignCurrencyTransactionGainLossBeforeTax |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionThe aggregate total of expenses of managing and administering the affairs of an entity, including affiliates of the reporting entity, which are not directly or indirectly associated with the manufacture, sale or creation of a product or product line. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-07(2)(a))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.5-03.4)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-2
| Name: |
us-gaap_GeneralAndAdministrativeExpense |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_IncomeStatementAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe portion of profit or loss for the period, net of income taxes, which is attributable to the parent. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482765/220-10-50-6
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-3
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (b)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-1
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-8
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 9
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-9
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-4
Reference 13: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-10
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section 45
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483581/946-220-45-7
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-04(18))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483586/944-220-S99-1
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-07(9))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-1
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(1)(d))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 20: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 21: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 22: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 23: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 24: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 25: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 26: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(B))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 27: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 28: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 29: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 30: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 31: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 60B
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-60B
Reference 32: http://www.xbrl.org/2003/role/exampleRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 31
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-31
Reference 33: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 34: http://www.xbrl.org/2003/role/disclosureRef
-Topic 205
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483499/205-20-50-7
Reference 35: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
Reference 36: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1A
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482790/220-10-45-1A
Reference 37: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1B
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482790/220-10-45-1B
Reference 38: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.5-03(20))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-2
Reference 39: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 942
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-04(22))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483589/942-220-S99-1
| Name: |
us-gaap_NetIncomeLoss |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionGenerally recurring costs associated with normal operations except for the portion of these expenses which can be clearly related to production and included in cost of sales or services. Includes selling, general and administrative expense.
| Name: |
us-gaap_OperatingExpenses |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_OperatingExpensesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe net result for the period of deducting operating expenses from operating revenues. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 4: http://www.xbrl.org/2003/role/exampleRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 31
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-31
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
| Name: |
us-gaap_OperatingIncomeLoss |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionThe aggregate costs incurred (1) in a planned search or critical investigation aimed at discovery of new knowledge with the hope that such knowledge will be useful in developing a new product or service, a new process or technique, or in bringing about a significant improvement to an existing product or process; or (2) to translate research findings or other knowledge into a plan or design for a new product or process or for a significant improvement to an existing product or process whether intended for sale or the entity's use, during the reporting period charged to research and development projects, including the costs of developing computer software up to the point in time of achieving technological feasibility, and costs allocated in accounting for a business combination to in-process projects deemed to have no alternative future use. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 730
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482916/730-10-50-1
Reference 2: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 912
-SubTopic 730
-Name Accounting Standards Codification
-Section 25
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482517/912-730-25-1
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 985
-SubTopic 20
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481283/985-20-50-1
| Name: |
us-gaap_ResearchAndDevelopmentExpense |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount, excluding tax collected from customer, of revenue from satisfaction of performance obligation by transferring promised good or service to customer. Tax collected from customer is tax assessed by governmental authority that is both imposed on and concurrent with specific revenue-producing transaction, including, but not limited to, sales, use, value added and excise. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 924
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SAB Topic 11.L)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479941/924-10-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 606
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 5
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479806/606-10-50-5
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 42
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-42
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 40
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-40
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 41
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-41
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 606
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479806/606-10-50-4
| Name: |
us-gaap_RevenueFromContractWithCustomerExcludingAssessedTax |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionThe average number of shares or units issued and outstanding that are used in calculating diluted EPS or earnings per unit (EPU), determined based on the timing of issuance of shares or units in the period. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 16
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-16
| Name: |
us-gaap_WeightedAverageNumberOfDilutedSharesOutstanding |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of [basic] shares or units, after adjustment for contingently issuable shares or units and other shares or units not deemed outstanding, determined by relating the portion of time within a reporting period that common shares or units have been outstanding to the total time in that period. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-10
| Name: |
us-gaap_WeightedAverageNumberOfSharesOutstandingBasic |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Consolidated Statements of Changes in Stockholders' Equity (Deficit) - USD ($)
|
Preferred Stock [Member] |
Common Stock [Member] |
Additional Paid-in Capital [Member] |
Retained Earnings [Member] |
Total |
| Balance at Sep. 30, 2021 |
|
$ 19
|
$ 83,949,046
|
$ (61,689,218)
|
$ 22,259,847
|
| Balance, shares at Sep. 30, 2021 |
|
195,579
|
|
|
|
| Sale of preferred stock and common stock warrants, net of issuance costs |
$ 1,540,640
|
|
597,574
|
|
2,138,214
|
| Sale of preferred stock and common stock warrants, net of issuance costs, shares |
22,500
|
|
|
|
|
| Conversion of preferred stock into common stock |
$ (1,540,640)
|
$ 3
|
1,540,637
|
|
|
| Conversion of preferred stock into common stock, shares |
(22,500)
|
25,101
|
|
|
|
| Sale of common stock, net of issuance costs |
|
$ 3
|
1,908,690
|
|
1,908,693
|
| Sale of common stock, net of issuance costs, shares |
|
30,206
|
|
|
|
| Issuance of common stock on vesting of restricted stock units |
|
|
|
|
|
| Issuance of common stock on vesting of restricted stock units, shares |
|
1,087
|
|
|
|
| Share-based compensation |
|
|
876,368
|
|
876,368
|
| Net loss |
|
|
|
(29,721,841)
|
(29,721,841)
|
| Balance at Sep. 30, 2022 |
|
$ 25
|
88,872,315
|
(91,411,059)
|
(2,538,719)
|
| Balance, shares at Sep. 30, 2022 |
|
251,973
|
|
|
|
| Sale of common stock, net of issuance costs |
|
$ 83
|
20,895,875
|
|
20,895,958
|
| Sale of common stock, net of issuance costs, shares |
|
833,287
|
|
|
|
| Issuance of common stock on vesting of restricted stock units |
|
$ 1
|
(1)
|
|
|
| Issuance of common stock on vesting of restricted stock units, shares |
|
7,676
|
|
|
|
| Share-based compensation |
|
|
248,626
|
|
248,626
|
| Net loss |
|
|
|
(18,832,694)
|
(18,832,694)
|
| Net share settlement of warrants |
|
$ 52
|
(52)
|
|
|
| Net share settlement of warrants, shares |
|
519,492
|
|
|
|
| Exercise of warrants |
|
$ 14
|
835
|
|
849
|
| Exercise of warrants, shares |
|
137,998
|
|
|
|
| Balance at Sep. 30, 2023 |
|
$ 175
|
$ 110,017,598
|
$ (110,243,753)
|
$ (225,980)
|
| Balance, shares at Sep. 30, 2023 |
|
1,750,426
|
|
|
|
| X |
- DefinitionWarrant exercises, shares.
| Name: |
SONN_SharesIssuedOnExerciseOfWarrantsShares |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionStock issued during period value net share settlement of warrants, shares.
| Name: |
SONN_StockIssuedDuringPeriodSharesNetShareSettlementOfWarrants |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionStock issued during period shares sale of preferred stock and common stock warrants
| Name: |
SONN_StockIssuedDuringPeriodSharesSaleOfPreferredStockAndCommonStockWarrants |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionStock issued during period value exercise of warrants.
| Name: |
SONN_StockIssuedDuringPeriodValueExerciseOfWarrants |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionStock issued during period value net share settlement of warrants.
| Name: |
SONN_StockIssuedDuringPeriodValueNetShareSettlementOfWarrants |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionStock issued during period value sale of preferred stock and common stock warrants.
| Name: |
SONN_StockIssuedDuringPeriodValueSaleOfPreferredStockAndCommonStockWarrants |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of increase to additional paid-in capital (APIC) for recognition of cost for award under share-based payment arrangement. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 35
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480483/718-10-35-2
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 718
-SubTopic 20
-Section 55
-Paragraph 13
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481089/718-20-55-13
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 718
-SubTopic 20
-Section 55
-Paragraph 12
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481089/718-20-55-12
| Name: |
us-gaap_AdjustmentsToAdditionalPaidInCapitalSharebasedCompensationRequisiteServicePeriodRecognitionValue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionThe portion of profit or loss for the period, net of income taxes, which is attributable to the parent. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482765/220-10-50-6
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-3
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (b)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-1
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-8
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 9
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-9
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-4
Reference 13: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-10
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section 45
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483581/946-220-45-7
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-04(18))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483586/944-220-S99-1
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-07(9))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-1
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(1)(d))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 20: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 21: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 22: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 23: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 24: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 25: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 26: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(B))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 27: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 28: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 29: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 30: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 31: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 60B
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-60B
Reference 32: http://www.xbrl.org/2003/role/exampleRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 31
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-31
Reference 33: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 34: http://www.xbrl.org/2003/role/disclosureRef
-Topic 205
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483499/205-20-50-7
Reference 35: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
Reference 36: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1A
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482790/220-10-45-1A
Reference 37: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1B
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482790/220-10-45-1B
Reference 38: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.5-03(20))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-2
Reference 39: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 942
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-04(22))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483589/942-220-S99-1
| Name: |
us-gaap_NetIncomeLoss |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of shares issued which are neither cancelled nor held in the treasury.
| Name: |
us-gaap_SharesOutstanding |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionNumber of shares issued during the period as a result of the conversion of convertible securities. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 1E
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481139/470-20-50-1E
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 4: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 505
-SubTopic 10
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-3
Reference 5: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.29-30)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_StockIssuedDuringPeriodSharesConversionOfConvertibleSecurities |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of new stock issued during the period. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 505
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481004/946-505-50-2
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(4)(b))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-03(i)(1))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479886/946-10-S99-3
Reference 5: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(28))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 6: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 7: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_StockIssuedDuringPeriodSharesNewIssues |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of shares issued during the period related to Restricted Stock Awards, net of any shares forfeited. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(28))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 4: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_StockIssuedDuringPeriodSharesRestrictedStockAwardNetOfForfeitures |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe gross value of stock issued during the period upon the conversion of convertible securities. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.29-31)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_StockIssuedDuringPeriodValueConversionOfConvertibleSecurities |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionEquity impact of the value of new stock issued during the period. Includes shares issued in an initial public offering or a secondary public offering. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 11
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-11
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 205
-Name Accounting Standards Codification
-Section 45
-Paragraph 4
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480767/946-205-45-4
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 505
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481004/946-505-50-2
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(4)(b))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 6: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(28))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 7: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 8: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_StockIssuedDuringPeriodValueNewIssues |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionValue of stock related to Restricted Stock Awards issued during the period, net of the stock value of such awards forfeited. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
| Name: |
us-gaap_StockIssuedDuringPeriodValueRestrictedStockAwardNetOfForfeitures |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of equity (deficit) attributable to parent. Excludes temporary equity and equity attributable to noncontrolling interest. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 2: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 12
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-12
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(19))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.6-05(4))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-2
Reference 5: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(4)(b))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(6))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(7))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 8: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 9: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 10: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 11: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 12: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(31))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 13: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(30))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 14: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 310
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SAB Topic 4.E)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480418/310-10-S99-2
| Name: |
us-gaap_StockholdersEquity |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
v3.23.3
Consolidated Statements of Cash Flows - USD ($)
|
12 Months Ended |
Sep. 30, 2023 |
Sep. 30, 2022 |
| Cash flows from operating activities: |
|
|
| Net loss |
$ (18,832,694)
|
$ (29,721,841)
|
| Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
| Acquired in-process research and development |
282,000
|
971,477
|
| Depreciation |
12,845
|
12,845
|
| Amortization of operating lease right-of-use asset |
62,905
|
80,412
|
| Share-based compensation |
248,626
|
876,368
|
| Changes in operating assets and liabilities: |
|
|
| Prepaid expenses and other current assets |
(33,653)
|
(454,269)
|
| Income tax receivable |
(69,269)
|
(717,305)
|
| Other assets |
(414,206)
|
|
| Accounts payable |
(2,631,215)
|
971,041
|
| Accrued expenses and other current liabilities |
231,953
|
641,372
|
| Operating lease liability |
(51,329)
|
(83,685)
|
| Deferred income |
(147,805)
|
(349,943)
|
| Net cash used in operating activities |
(21,341,842)
|
(27,773,528)
|
| Cash flows from investing activities: |
|
|
| Purchases of in-process research and development |
(443,250)
|
(810,227)
|
| Net cash used in investing activities |
(443,250)
|
(810,227)
|
| Cash flows from financing activities: |
|
|
| Proceeds from sale of preferred stock and common stock warrants, net of issuance costs |
|
2,172,649
|
| Proceeds from sale of common stock, net of issuance costs |
21,006,371
|
1,841,918
|
| Proceeds from exercise of warrants, net of issuance costs |
849
|
|
| Repayments of related party notes |
(748)
|
|
| Net cash provided by financing activities |
21,006,472
|
4,014,567
|
| Net decrease in cash |
(778,620)
|
(24,569,188)
|
| Cash, beginning of year |
3,052,879
|
27,622,067
|
| Cash, end of year |
2,274,259
|
3,052,879
|
| Supplemental disclosure of non-cash operating, investing and financing activities: |
|
|
| Change in operating lease right-of-use asset and liability due to amended lease |
|
213,793
|
| Deferred offering costs charged against proceeds from sale of common stock |
32,340
|
|
| Deferred offering costs in accounts payable and accrued expenses |
49,988
|
80,940
|
| Net settlement of warrants |
52
|
|
| In-process research and development in accrued expenses |
|
161,250
|
| Common stock issuance costs in accounts payable and accrued expenses |
$ 78,073
|
|
| X |
- DefinitionThe amount of purchased research and development assets that are acquired in a business combination have no alternative future use and are therefore written off in the period of acquisition.
| Name: |
SONN_AcquiredInprocessResearchAndDevelopment |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionChange in operating right of use asset and liability due to amended lease.
| Name: |
SONN_ChangeInOperatingRightOfUseAssetAndLiabilityDueToAmendedLease |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionCommon stock issuance costs in accounts payable and accrued expenses.
| Name: |
SONN_CommonStockIssuanceCostsInAccountsPayableAndAccruedExpenses |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionDeferred offering costs charged against proceeds from sale of common stock.
| Name: |
SONN_DeferredOfferingCostsChargedAgainstProceedsFromSaleOfCommonStock |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionDeferred offering costs in accounts payable and accrued expenses.
| Name: |
SONN_DeferredOfferingCostsInAccountsPayableAndAccruedExpenses |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionIn process research and development in accrued expenses.
| Name: |
SONN_InProcessResearchAndDevelopmentInAccruedExpenses |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionThe value of net settlement of warrants.
| Name: |
SONN_NetSettlementOfWarrants |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_AdjustmentsToReconcileNetIncomeLossToCashProvidedByUsedInOperatingActivitiesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of cash and cash equivalents, and cash and cash equivalents restricted to withdrawal or usage; including, but not limited to, disposal group and discontinued operations. Cash includes, but is not limited to, currency on hand, demand deposits with banks or financial institutions, and other accounts with general characteristics of demand deposits. Cash equivalents include, but are not limited to, short-term, highly liquid investments that are both readily convertible to known amounts of cash and so near their maturity that they present insignificant risk of changes in value because of changes in interest rates. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482913/230-10-50-8
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 24
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-24
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 230
-SubTopic 10
-Section 45
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-4
| Name: |
us-gaap_CashCashEquivalentsRestrictedCashAndRestrictedCashEquivalentsIncludingDisposalGroupAndDiscontinuedOperations |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of increase (decrease) in cash, cash equivalents, and cash and cash equivalents restricted to withdrawal or usage; including effect from exchange rate change. Cash includes, but is not limited to, currency on hand, demand deposits with banks or financial institutions, and other accounts with general characteristics of demand deposits. Cash equivalents include, but are not limited to, short-term, highly liquid investments that are both readily convertible to known amounts of cash and so near their maturity that they present insignificant risk of changes in value because of changes in interest rates. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 24
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-24
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-SubTopic 230
-Topic 830
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481877/830-230-45-1
| Name: |
us-gaap_CashCashEquivalentsRestrictedCashAndRestrictedCashEquivalentsPeriodIncreaseDecreaseIncludingExchangeRateEffect |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe amount of expense recognized in the current period that reflects the allocation of the cost of tangible assets over the assets' useful lives. Includes production and non-production related depreciation. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (b)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 360
-SubTopic 10
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482099/360-10-50-1
| Name: |
us-gaap_Depreciation |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe increase (decrease) during the reporting period in the aggregate amount of liabilities incurred (and for which invoices have typically been received) and payable to vendors for goods and services received that are used in an entity's business. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
| Name: |
us-gaap_IncreaseDecreaseInAccountsPayable |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe increase (decrease) during the reporting period in the aggregate amount of expenses incurred but not yet paid. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
| Name: |
us-gaap_IncreaseDecreaseInAccruedLiabilities |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of increase (decrease) in obligation to transfer good or service to customer for which consideration has been received or is receivable. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 912
-SubTopic 310
-Name Accounting Standards Codification
-Section 45
-Paragraph 11
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482312/912-310-45-11
Reference 2: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
| Name: |
us-gaap_IncreaseDecreaseInContractWithCustomerLiability |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_IncreaseDecreaseInOperatingCapitalAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of increase (decrease) in operating assets classified as other. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
| Name: |
us-gaap_IncreaseDecreaseInOtherOperatingAssets |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of increase (decrease) in operating liabilities classified as other. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
| Name: |
us-gaap_IncreaseDecreaseInOtherOperatingLiabilities |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of increase (decrease) in prepaid expenses, and assets classified as other. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
| Name: |
us-gaap_IncreaseDecreaseInPrepaidDeferredExpenseAndOtherAssets |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of cash inflow (outflow) from financing activities, including discontinued operations. Financing activity cash flows include obtaining resources from owners and providing them with a return on, and a return of, their investment; borrowing money and repaying amounts borrowed, or settling the obligation; and obtaining and paying for other resources obtained from creditors on long-term credit. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 24
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-24
| Name: |
us-gaap_NetCashProvidedByUsedInFinancingActivities |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_NetCashProvidedByUsedInFinancingActivitiesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of cash inflow (outflow) from investing activities, including discontinued operations. Investing activity cash flows include making and collecting loans and acquiring and disposing of debt or equity instruments and property, plant, and equipment and other productive assets. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 24
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-24
| Name: |
us-gaap_NetCashProvidedByUsedInInvestingActivities |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_NetCashProvidedByUsedInInvestingActivitiesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of cash inflow (outflow) from operating activities, including discontinued operations. Operating activity cash flows include transactions, adjustments, and changes in value not defined as investing or financing activities. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 24
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-24
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 25
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-25
| Name: |
us-gaap_NetCashProvidedByUsedInOperatingActivities |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_NetCashProvidedByUsedInOperatingActivitiesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe portion of profit or loss for the period, net of income taxes, which is attributable to the parent. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(g)(1)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 323
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481687/323-10-50-3
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 825
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 28
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-28
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482765/220-10-50-6
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-3
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (b)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-1
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 65
-Paragraph 1
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480175/815-40-65-1
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-8
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 9
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-9
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-11
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 250
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483443/250-10-50-4
Reference 13: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-10
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section 45
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483581/946-220-45-7
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-04(18))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483586/944-220-S99-1
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-07(9))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-1
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(1)(d))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 20: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(ii))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 21: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 22: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 23: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1A
-Subparagraph (SX 210.13-01(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1A
Reference 24: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(i))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 25: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(A))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 26: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iii)(B))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 27: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(4)(iv))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 28: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1B
-Subparagraph (SX 210.13-02(a)(5))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480097/470-10-S99-1B
Reference 29: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 30: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (f)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 31: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 60B
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482689/260-10-45-60B
Reference 32: http://www.xbrl.org/2003/role/exampleRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 31
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-31
Reference 33: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 34: http://www.xbrl.org/2003/role/disclosureRef
-Topic 205
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 7
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483499/205-20-50-7
Reference 35: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
Reference 36: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1A
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482790/220-10-45-1A
Reference 37: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 1B
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482790/220-10-45-1B
Reference 38: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.5-03(20))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483621/220-10-S99-2
Reference 39: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 942
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.9-04(22))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483589/942-220-S99-1
| Name: |
us-gaap_NetIncomeLoss |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of periodic reduction over lease term of carrying amount of right-of-use asset from operating lease. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
| Name: |
us-gaap_OperatingLeaseRightOfUseAssetAmortizationExpense |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe cash outflows from the purchase of net carrying value allocated to in-process research and development costs and materials acquired in a business combination. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 230
-SubTopic 10
-Section 45
-Paragraph 13
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-13
| Name: |
us-gaap_PaymentsToAcquireInProcessResearchAndDevelopment |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionThe cash inflow from the additional capital contribution to the entity. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 14
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-14
| Name: |
us-gaap_ProceedsFromIssuanceOfCommonStock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionProceeds from issuance of capital stock which provides for a specific dividend that is paid to the shareholders before any dividends to common stockholders and which takes precedence over common stockholders in the event of liquidation. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 14
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-14
| Name: |
us-gaap_ProceedsFromIssuanceOfPreferredStockAndPreferenceStock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe cash inflow associated with the amount received from holders exercising their stock warrants. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 230
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 14
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-14
| Name: |
us-gaap_ProceedsFromWarrantExercises |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of noncash expense for share-based payment arrangement. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
| Name: |
us-gaap_ShareBasedCompensation |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
v3.23.3
Organization and Description of Business
|
12 Months Ended |
Sep. 30, 2023 |
|---|
| Organization, Consolidation and Presentation of Financial Statements [Abstract] |
|
| Organization and Description of Business |
1.
Organization and Description of Business
Description
of business
Sonnet
BioTherapeutics, Inc. (“Prior Sonnet”) was incorporated as a New Jersey corporation on April 6, 2015. Prior Sonnet completed
a merger with publicly-held Chanticleer Holdings, Inc. (“Chanticleer”) on April 1, 2020. After the merger, Chanticleer changed
its name to Sonnet BioTherapeutics Holdings, Inc. (“Sonnet” or the “Company”). Sonnet is a clinical stage, oncology-focused
biotechnology company with a proprietary platform for innovating biologic medicines of single or bifunctional action. Known as FHAB™
(Fully Human Albumin Binding), the technology utilizes a fully human single chain antibody fragment (scFv) that binds to and “hitch-hikes”
on human serum albumin (“HSA”) for transport to target tissues.
Sonnet’s
lead proprietary asset, SON-1010, is a fully human version of Interleukin 12 (“IL-12”), covalently linked to the FHAB
construct, for which Sonnet is pursuing clinical development in solid tumor indications, including ovarian cancer, non-small cell
lung cancer and head and neck cancer. In March 2022, the FDA cleared Sonnet’s Investigational New Drug (“IND”) application
for SON-1010. This allowed the Company to initiate a U.S. clinical trial (SB101) in oncology patients with solid tumors during the second
calendar quarter of 2022. In September 2021, the Company created a wholly-owned Australian subsidiary, SonnetBio Pty Ltd (“Subsidiary”),
for the purpose of conducting certain clinical trials. Sonnet received approval and initiated an Australian clinical study (SB102) of
SON-1010 in healthy volunteers during the third calendar quarter of 2022. Interim safety and tolerability data from the SB101 and SB102
studies were reported in April 2023.
In
January 2023, Sonnet announced a collaboration agreement with Roche for the clinical evaluation of SON-1010 with atezolizumab (Tecentriq®).
The companies have entered into a Master Clinical Trial and Supply Agreement (“MCSA”), along with ancillary Quality and Safety
Agreements, to study the safety and efficacy of the combination of SON-1010 and atezolizumab in a platinum-resistant ovarian cancer (“PROC”)
patient setting. Further, the companies will provide SON-1010 and atezolizumab, respectively, for use in the Phase 1b/Phase 2a combination
safety, dose-escalation, and efficacy study (SB221). Part 1 of this 2-part study was approved in June 2023 by the local Human
Research Ethics Committee in Australia under CT-2023-CTN-01399-1 and the Therapeutic Goods Administration has been notified. In August
2023, the FDA accepted the IND for SB221. The trial consists of a modified 3+3 dose-escalation design in Part 1 to establish the maximum
tolerated dose (MTD) of SON-1010 with a fixed dose of atezolizumab. Clinical benefit in PROC will be confirmed in an expansion group to
establish the recommended Phase 2 dose (RP2D). Part 2 of the study will then investigate SON-1010 monotherapy, its use in combination
with atezolizumab, or the standard of care (SOC) for PROC in a randomized comparison to show proof-of-concept (POC).
As part of the ongoing cost-cutting evaluations, all antiviral development with SON-1010 has been suspended.
The Company acquired the global development rights to its most advanced
compound, SON-080, a fully human version of Interleukin 6 (“IL-6”), in April 2020 through its acquisition of the outstanding
shares of Relief Therapeutics SA. Sonnet is advancing SON-080 in target indications of Chemotherapy-Induced Peripheral Neuropathy (“CIPN”)
and Diabetic Peripheral Neuropathy (“DPN”). Sonnet received approval to initiate an ex-U.S. Phase 1b/2a study with SON-080
in CIPN during the third quarter of 2022. The Data Safety Monitoring Board (“DSMB”) overseeing the study is expected to meet
during the first calendar quarter of 2024. Following the completion of the DSMB review, Sonnet anticipates announcing initial safety data
from the CIPN study. Pursuant to a license agreement the Company entered into with New Life Therapeutics Pte, Ltd. (“New Life”)
of Singapore in May 2021, Sonnet and New Life will be jointly responsible for developing SON-080 in DPN. The objective will be to analyze
the data and to consider initiating a Phase 2 study once the CIPN safety data has been evaluated.
SON-1210
(IL12-FHAB-IL15), Sonnet’s lead bi-specific construct, combines FHAB with fully human IL-12 and fully human
Interleukin 15 (“IL-15”). This compound is being developed for solid tumor indications, including colorectal cancer. In February
2023, the Company announced the successful completion of two IND-enabling toxicology studies with SON-1210 in non-human primates. Sonnet
is prepared to initiate the regulatory authorization process for SON-1210 pending the outcome of any partnering activity.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
SON-1410
(IL18-FHAB-IL12) is a bi-specific combination of Interleukin 18 (“IL-18”) and IL-12 for solid tumor cancers. Cell line development and process development are ongoing, with early experimental
drug supply suitable for formulation and analytical method development activities. After some delays in 2023, activities will continue
into 2024 with the potential to generate a drug suitable for preclinical studies and subsequent human studies.
The
Company has completed sequence confirmation for SON-3015 (anti-IL6-FHAB-anti-TGFβ). Early stage bi-specific drug has
been generated and is being stored for future use in in vivo mice studies. Sonnet has elected to place the SON-3015 development
program on hold for expense reduction purposes.
Liquidity
The
Company has incurred recurring losses and negative cash flows from operations since inception and it expects to generate losses from
operations for the foreseeable future primarily due to research and development costs for its potential product candidates. The Company
believes its cash of $2.3 million at September 30, 2023, in addition to the $4.1 million in net proceeds received from the October Offering
described below, will fund the Company’s projected operations into March 2024. The Company also expects to receive a $0.8 million net cash refund from
the research and development tax incentive program in Australia (see Note 2) and recently received preliminary approval of its application
to sell up to $4.8 million of its New Jersey state net operating losses through the Technology Business Tax Certificate Transfer Program
(the “Program”), subject to execution of such sale (see Note 10). Substantial additional financing will be needed
by the Company to fund its operations. These factors raise substantial doubt about the Company’s ability to continue as a going
concern. The accompanying consolidated financial statements have been prepared on a going concern basis, which contemplates the realization
of assets and satisfaction of liabilities in the normal course of business. The consolidated financial statements do not include any
adjustments that might result from the outcome of this uncertainty.
On
October 26, 2023, the Company closed a public offering of common stock and certain warrants through Chardan Capital Markets, LLC and
Ladenburg Thalmann & Co. Inc. as underwriters, for net proceeds of $4.1 million through the issuance and sale of 1,306,250 shares
of its common stock and, to certain investors, pre-funded warrants to purchase 1,537,500 shares of common stock, and accompanying common
warrants to purchase up to an aggregate of 5,687,500 shares of its common stock (the “October Offering”). Each share of common
stock and pre-funded warrant to purchase one share of common stock was sold together with a common warrant to purchase two shares of
common stock. The public offering price of each share of common stock and accompanying common warrant was $1.60 and the public offering
price of each pre-funded warrant and accompanying common warrant was $1.5999. The common warrants are immediately exercisable at a price
of $1.60 per share of common stock, expire five years from the date of issuance and contain an alternative cashless exercise provision.
The pre-funded warrants are immediately exercisable at any time, until exercised in full, at a price of $0.0001 per share of common stock.
In addition, warrants to purchase 85,312 shares of common stock were issued to the underwriters as compensation for their services related
to the offering. These common stock warrants have an exercise price of $2.00 per share and expire five years from the date of issuance.
The
Company plans to secure additional capital in the future through equity or debt financings, partnerships, collaborations, or other sources
to carry out the Company’s planned development activities. If additional capital is not available when required, the Company may
need to delay or curtail its operations until such funding is received. Various internal and external factors will affect whether and
when the Company’s product candidates become approved for marketing and successful commercialization. The regulatory approval and
market acceptance of the Company’s product candidates, length of time and cost of developing and commercializing these product
candidates and/or failure of them at any stage of the approval process will materially affect the Company’s financial condition
and future operations.
Operations
since inception have consisted primarily of organizing the Company, securing financing, developing technologies through research and
development and conducting preclinical studies. The Company faces risks associated with companies whose products are in development.
These risks include the need for additional financing to complete its research and development, achieving its research and development
objectives, defending its intellectual property rights, recruiting and retaining skilled personnel, and dependence on key members of
management.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
|
| X |
- DefinitionThe entire disclosure for the nature of an entity's business, major products or services, principal markets including location, and the relative importance of its operations in each business and the basis for the determination, including but not limited to, assets, revenues, or earnings. For an entity that has not commenced principal operations, disclosures about the risks and uncertainties related to the activities in which the entity is currently engaged and an understanding of what those activities are being directed toward. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Topic 275
-Publisher FASB
-URI https://asc.fasb.org//275/tableOfContent
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-SubTopic 10
-Topic 275
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482861/275-10-50-1
| Name: |
us-gaap_NatureOfOperations |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_OrganizationConsolidationAndPresentationOfFinancialStatementsAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Summary of Significant Accounting Policies
|
12 Months Ended |
Sep. 30, 2023 |
|---|
| Accounting Policies [Abstract] |
|
| Summary of Significant Accounting Policies |
2.
Summary of Significant Accounting Policies
a.
Basis of presentation
The
accompanying consolidated financial statements have been prepared in conformity with U.S. generally accepted accounting principles (“U.S.
GAAP”). Any reference in these notes to applicable guidance is meant to refer to U.S. GAAP as found in the Accounting Standards
Codification (“ASC”) and Accounting Standards Updates (“ASU”) promulgated by the Financial Accounting Standards
Board (“FASB”).
b.
Consolidation
The
consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries. All intercompany accounts and
transactions have been eliminated in consolidation.
c.
Use of estimates
The
preparation of the consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions
that affect the amounts reported in the consolidated financial statements and accompanying notes. Significant estimates and assumptions
reflected in these consolidated financial statements include the accrual of research and development expenses. Estimates and assumptions
are periodically reviewed in-light of changes in circumstances, facts and experience. Changes in estimates are recorded in the period
in which they become known. Actual results could differ from management’s estimates.
d.
Reclassifications
Certain
amounts from the prior period have been reclassified to conform with the current period presentation.
e.
Segment information
Operating
segments are defined as components of an enterprise about which separate discrete information is available for evaluation by the chief
operating decision maker, or decision-making group, in deciding how to allocate resources and assess performance. The Company views its
operations and manages its business in one segment.
f.
Fair value of financial instruments
Management
believes that the carrying amounts of the Company’s financial instruments, including accounts payable, approximate fair value due
to the short-term nature of those instruments.
g.
Property and equipment
Property
and equipment are recorded at cost and depreciated using the straight-line method over the estimated useful lives of the assets. Expenditures
for repairs and maintenance that do not extend the estimated useful life or improve an asset are expensed as incurred. Upon retirement
or sale, the cost and related accumulated depreciation and amortization of assets disposed of are removed from the accounts, and any
resulting gain or loss is included in the consolidated statement of operations.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
h.
Impairment of long-lived assets
The
Company reviews long-lived assets, such as property and equipment, for impairment whenever events or changes in circumstances indicate
that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison
of the carrying amount of an asset to the undiscounted future cash flows expected to be generated by that asset. If the carrying amount
of an asset exceeds its estimated undiscounted future cash flows, then an impairment charge is recognized for the amount by which the
carrying value of the asset exceeds the estimated fair value of the asset. There were no impairment charges recorded during the fiscal
years ended September 30, 2023 and 2022.
i.
Deferred offering costs
Legal
and other costs incurred in relation to equity offerings are capitalized as deferred offering costs and charged against the proceeds
from equity offerings when received. If a financing is abandoned, deferred offering costs are expensed. As of September 30, 2023, the
Company had $49,988 in deferred offering costs associated with the October Offering. As of September 30, 2022, the Company had $0.1 million
in deferred offering costs associated with the 2022 Sales Agreement (see Note 7).
j.
Incentive tax receivable
Subsidiary
is eligible to participate in an Australian research and development tax incentive program. As part of this program, Subsidiary is eligible
to receive a cash refund from the Australian Taxation Office for a percentage of the research and development costs expended by Subsidiary
in Australia. The
cash refund is available to eligible companies with annual aggregate revenues of less than $20.0 million (Australian) during the reimbursable
period. The Company estimates the amount of cash
refund it expects to receive related to the Australian research and development tax incentive program and records the incentive when
it is probable (i) the Company will comply with relevant conditions of the program and (ii) the incentive will be received. As of both
September 30, 2023 and 2022, the Company’s estimate of the amount of cash refund it expects to receive for eligible spending related
to the Australian research and development tax incentive program was $0.8
million. For each of the years ended September
30, 2023 and 2022, $0.8
million for the expected net cash refund related to the tax
incentive program was included in research and development expenses. In January 2023, the Company received $1.1
million from the Australian government related
to eligible research and development expenses for the year ended September 30, 2022. In April 2023, the Company refunded the Australian
government for $0.2 million
due to new information on the Company’s clinical trial studies.
k.
Collaboration revenue
Collaboration
arrangements may contain multiple components, which may include (i) licenses; (ii) research and development activities; and (iii) the
manufacturing and supply of certain materials. Payments pursuant to these arrangements may include non-refundable payments, upfront payments,
milestone payments upon the achievement of significant regulatory and development events, sales milestones and royalties on product sales.
The amount of variable consideration is constrained until it is probable that the revenue is not at a significant risk of reversal in
a future period.
In
determining the appropriate amount of revenue to be recognized as the Company fulfills its obligations under a collaboration arrangement,
the Company performs the following steps: (i) identification of the promised goods or services in the contract; (ii) determination of
whether the promised goods or services are performance obligations, including whether they are capable of being distinct; (iii) measurement
of the transaction price, including the constraint on variable consideration; (iv) allocation of the transaction price to the performance
obligations; and (v) recognition of revenue as the Company satisfies each performance obligation.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
The
Company applies significant judgment when evaluating whether contractual obligations represent distinct performance obligations, allocating
transaction price to performance obligations within a contract, determining when performance obligations have been met, and assessing
the recognition of variable consideration. When consideration is received prior to the Company completing its performance obligation
under the terms of a contract, a contract liability is recorded as deferred income. Deferred income expected to be recognized as revenue
within the twelve months following the balance sheet date is classified as a current liability. In May 2021, the Company entered into
a License Agreement (the “New Life Agreement”) with New Life. See Note 6 for further discussion of the New Life Agreement.
l.
Research and development expense
Research
and development expenses include all direct and indirect costs associated with the development of the Company’s biopharmaceutical
products. These expenses include personnel costs, consulting fees, and payments to third parties for research, development, and manufacturing
services. These costs are charged to expense as incurred.
At
the end of the reporting period, the Company compares payments made to third-party service providers to the estimated progress toward
completion of the related project, based on the measure of progress as defined in the contract. Factors the Company considers in preparing
the estimates include costs incurred by the service provider, milestones achieved, and other criteria related to the efforts of its service
providers. Such estimates are subject to change as additional information becomes available. Depending on the timing of payment to the
service providers and the progress that the Company estimates has been made as a result of the service provided, the Company will record
a prepaid expense or accrued liability relating to these costs. Upfront milestone payments made to third parties who perform research
and development services on the Company’s behalf are expensed as services are rendered. Contingent development or regulatory milestone
payments are recognized upon the related resolution of such contingencies.
m.
Foreign currency
Transaction
gains and losses resulting from exchange rate changes on transactions denominated in currencies other than the U.S. dollar are included
in operations in the period in which the transaction occurs and reported within the foreign exchange loss line item in the consolidated
statements of operations.
n.
Share-based compensation
The
Company measures equity classified share-based awards granted to employees and nonemployees based on the estimated fair value on the
date of grant and recognizes compensation expense of those awards over the requisite service period, which is the vesting period of the
respective award. The Company accounts for forfeitures as they occur. For share-based awards with service-based vesting conditions, the
Company recognizes compensation expense on a straight-line basis over the service period. The Company classifies share-based compensation
expense in its consolidated statements of operations in the same manner in which the award recipient’s payroll costs are classified
or in which the award recipient’s service payments are classified.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
o.
Income taxes
The
Company uses the asset-and-liability method of accounting for income taxes. Deferred tax assets and liabilities are recognized for the
estimated future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and
liabilities and their respective tax bases, and operating loss and credit carryforwards. Deferred tax assets and liabilities are measured
using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered
or settled. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not
be realized. The Company recognizes the benefit of an uncertain tax position that it has taken or expects to take on its income tax return
if such a position is more likely than not to be sustained.
p.
Reverse stock split
On
August 31, 2023, the Company filed a Certificate of Amendment to its Certificate of Incorporation, as amended, with the Secretary of
State of the State of Delaware, which effected a 1-for
-22 reverse stock split of the Company’s issued and outstanding shares of common stock. As a result of the reverse
stock split, every 22 shares of common stock issued and outstanding was converted into one share of common stock. The reverse stock
split affected all stockholders uniformly and did not alter any stockholder’s percentage interest in the Company’s
equity. No fractional shares were issued in connection with the reverse stock split. Stockholders who would otherwise be entitled to
a fractional share of common stock were instead entitled to receive a proportional cash payment. The reverse stock split did not
change the par value or authorized number of shares of common stock. All common share and per share amounts presented in the
consolidated financial statements and accompanying notes have been retroactively adjusted to reflect the reverse stock
split.
q.
Net loss per share
Basic
net loss per share is computed by dividing net loss by the weighted-average number of shares of common stock outstanding during each
period (and potential shares of common stock that are exercisable for little or no consideration).
Diluted
loss per share includes the effect, if any, from the potential exercise or conversion of securities such as common stock warrants and
stock options which would result in the issuance of incremental shares of common stock. For diluted net loss per share, the weighted-average
number of shares of common stock is the same for basic net loss per share due to the fact that when a net loss exists, dilutive securities
are not included in the calculation as the impact is anti-dilutive.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
The
following potentially dilutive securities have been excluded from the computation of diluted shares of common stock outstanding as they
would be anti-dilutive:
Schedule of Potentially Dilutive Securities
| | |
2023 | | |
2022 | |
| | |
September
30, | |
| | |
2023 | | |
2022 | |
| Common
stock warrants August 2021 | |
| 128,500 | | |
| 128,500 | |
| Underwriter
warrants August 2021 | |
| 2,287 | | |
| 2,287 | |
| Private
warrants | |
| — | | |
| 332 | |
| Chanticleer
warrants | |
| 57 | | |
| 57 | |
| Series
C warrants | |
| 36,778 | | |
| 36,778 | |
| Series
3 warrants | |
| 12,548 | | |
| 12,548 | |
| Unvested
restricted stock units and awards | |
| 2,326 | | |
| 2,162 | |
| Common
stock warrants February 2023 | |
| 271,883 | | |
| — | |
| Underwriter
warrants February 2023 | |
| 44,190 | | |
| — | |
| Common
stock private placement warrants June 2023 | |
| 227,272 | | |
| — | |
| Placement
agent warrants June 2023 | |
| 6,818 | | |
| — | |
| Total
anti-dilutive weighted average shares | |
| 732,659 | | |
| 182,664 | |
r.
Recent accounting pronouncements
Recently
adopted
In
May 2021, the FASB issued ASU 2021-04, Earnings Per Share (Topic 260), Debt-Modifications and Extinguishments (Subtopic 470-50), Compensation-Stock
Compensation (Topic 718), and Derivatives and Hedging-Contracts in Entity’s Own Equity (Subtopic 815-40): Issuer’s Accounting
for Certain Modifications or Exchanges of Freestanding Equity-Classified Written Call Options. The amendments in ASU 2021-04 provide
guidance to clarify and reduce diversity in an issuer’s accounting for modifications or exchanges of freestanding equity-classified
written call options (for example, warrants) that remain equity classified after modification or exchange. The adoption of ASU 2021-04
on October 1, 2022 did not have any impact on the consolidated financial statements.
In
November 2021, the Financial Accounting Standards Board, or FASB, issued Accounting Standards Update No. 2021-10, Government Assistance
(Topic 832): Disclosure by Business Entities about Government Assistance, or ASU 2021-10, which improves the transparency of government
assistance received by most business entities by requiring the disclosure of: (1) the types of government assistance received; (2) the
accounting for such assistance; and (3) the effect of the assistance on a business entity’s financial statements. This guidance
is effective for financial statements issued for annual periods beginning after 15 December 2021. The adoption of the guidance on October
1, 2022 did not have a material impact on our consolidated financial statements.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
|
| X |
- References
+ Details
| Name: |
us-gaap_AccountingPoliciesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe entire disclosure for all significant accounting policies of the reporting entity. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483426/235-10-50-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 235
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//235/tableOfContent
| Name: |
us-gaap_SignificantAccountingPoliciesTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Accrued Expenses and Other Current Liabilities
|
12 Months Ended |
Sep. 30, 2023 |
|---|
| Payables and Accruals [Abstract] |
|
| Accrued Expenses and Other Current Liabilities |
3.
Accrued Expenses and Other Current Liabilities
Accrued
expenses and other current liabilities consisted of the following:
Schedule
of Accrued Expenses and Other Current Liabilities
| | |
2023 | | |
2022 | |
| | |
September
30, | |
| | |
2023 | | |
2022 | |
| Compensation
and benefits | |
$ | 2,091,196 | | |
$ | 1,218,530 | |
| Research
and development | |
| 913,145 | | |
| 1,593,922 | |
| Professional
fees | |
| 224,031 | | |
| 378,890 | |
| Other | |
| 2,550 | | |
| 2,630 | |
| Accrued
expenses and other current liabilities | |
$ | 3,230,922 | | |
$ | 3,193,972 | |
|
| X |
- DefinitionThe entire disclosure for accounts payable, accrued expenses, and other liabilities that are classified as current at the end of the reporting period.
| Name: |
us-gaap_AccountsPayableAccruedLiabilitiesAndOtherLiabilitiesDisclosureCurrentTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_PayablesAndAccrualsAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Leases
|
12 Months Ended |
Sep. 30, 2023 |
|---|
| Leases [Abstract] |
|
| Leases |
4.
Leases
In
December 2019, the Company entered into a 36-month lease for office space in Princeton, New Jersey, which commenced February 1, 2020.
In May 2022, the Company amended the existing lease agreement in order to increase the lease term by approximately three years, which
has been accounted for as a lease modification. The operating lease right-of-use asset and liability were remeasured at the modification
date, resulting in an increase to both balances of $0.2 million.
The
components of lease expense for the years ended September 30, 2023 and 2022 are as follows:
Schedule
of Lease Expenses
| Lease
expense | |
2023 | | |
2022 | |
| Operating
lease expense | |
$ | 90,837 | | |
$ | 94,828 | |
| Variable
lease expense | |
| 5,978 | | |
| 5,339 | |
| Total
lease cost | |
$ | 96,815 | | |
$ | 100,167 | |
At
September 30, 2023, the weighted average remaining lease term was 2.6 years and the weighted average discount rate was 12%.
Cash
flow information related to operating leases for the years ended September 30, 2023 and 2022 is as follows:
Schedule of Operating Lease Liabilities
| Cash
paid for amounts included in the measurement of lease liabilities: | |
2023 | | |
2022 | |
| Operating
cash flows from operating leases | |
$ | 79,259 | | |
$ | 98,101 | |
Future
minimum lease payments under non-cancellable leases at September 30, 2023 are as follows:
Schedule of Future Minimum Lease Payments
| | |
| | |
| Fiscal
year | |
| | |
| 2024 | |
$ | 93,614 | |
| 2025 | |
| 95,487 | |
| 2026 | |
| 48,216 | |
| Total
undiscounted lease payments | |
| 237,317 | |
| Less:
imputed interest | |
| (33,406 | ) |
| Total
lease liabilities | |
$ | 203,911 | |
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
|
| X |
- References
+ Details
| Name: |
us-gaap_LeasesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe entire disclosure for operating leases of lessee. Includes, but is not limited to, description of operating lease and maturity analysis of operating lease liability. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//842-20/tableOfContent
| Name: |
us-gaap_LesseeOperatingLeasesTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Commitments and Contingencies
|
12 Months Ended |
Sep. 30, 2023 |
|---|
| Commitments and Contingencies Disclosure [Abstract] |
|
| Commitments and Contingencies |
5.
Commitments and Contingencies
Legal
proceedings
From
time to time, the Company is a party to various lawsuits, claims, and other legal proceedings that arise in the ordinary course of its
business. While the outcomes of these matters are uncertain, management does not expect that the ultimate costs to resolve these matters
will have a material adverse effect on the Company’s consolidated financial position, results of operations, or cash flows.
License
agreements
In
July 2012, the Company entered into a Discovery Collaboration Agreement (the “Collaboration Agreement”) with XOMA (US) LLC
(“XOMA”), pursuant to which XOMA granted to the Company a non-exclusive, non-transferable license and/or right to use certain
materials, technologies and related information related to discovery, optimization and development of antibodies and related proteins
and to develop and commercialize products thereunder. The Company is obligated to make contingent milestone payments to XOMA totaling
$3.8 million on a product-by-product basis upon the achievement of certain development and approval milestones related to a product.
The Company has also agreed to pay XOMA low single-digit royalties on net sales of products sold by the Company. Royalties on each product
are payable on a country-by-country basis until the later of (i) a specified period of time after the first commercial sale, and (ii)
the date of expiration of the last valid claim in the last-to-expire of the issued patents covered by the Collaboration Agreement. The
first milestone was achieved in April 2022, at which time the Company incurred a $0.5 million license fee which was recorded as acquired
in-process research and development and included as research and development expenses in the consolidated statement of operations for
the year ended September 30, 2022. No license fees were incurred during the year ended September 30, 2023.
In
August 2015, the Company entered into a License Agreement (the “ARES License Agreement”) with Ares Trading, a wholly-owned
subsidiary of Merck KGaA (“ARES”). Under the terms of the ARES License Agreement, ARES has granted the Company a sublicensable,
exclusive, worldwide, royalty-bearing license on proprietary patents to research, develop, use and commercialize products using atexakin
alfa (“Atexakin”), a low dose formulation of human IL-6 in peripheral neuropathies and vascular complications. Pursuant to
the ARES License Agreement, the Company will pay ARES high single-digit royalties on net sales of products sold by the Company. Royalties
are payable on a product-by-product and country-by-country basis until the later of (i) a specified period of time after the first commercial
sale in such country, and (ii) the last date on which such product is covered by a valid claim in such country.
In
January 2019, the Company entered into a Frame Services and License Agreement (the “Cellca Agreement”) with Sartorius Stedim
Cellca GMBH (“Cellca”), pursuant to which Cellca has granted the Company a worldwide, non-exclusive, perpetual, non-transferable
license to develop, manufacture or have manufactured, use, sell, import, export and/or otherwise commercialize product based on Cellca’s
work to generate a specified transfected cell line and develop an upstream production process for such cell line. The Cellca Agreement
is effective unless terminated by either party by giving six months notice, or by giving 14 days notice if terminated for good cause.
The Company is obligated to make milestone payments to Cellca totaling up to $0.7 million upon the achievement of certain development
and approval milestones if the Buy-Out Option is not exercised. The Company has a Buy-Out Option that will be effective between the time
of completion of a clinical trial and the receipt of regulatory approval for commercialization of product. The cost to exercise the Buy-Out
Option increases on each anniversary of the commencement date of the Buy-Out Option Period, and ranges from $0.1 million to $0.6 million.
The cost to exercise the Buy-Out Option will replace the $0.6 million contingent milestone payment due upon final regulatory approval.
The first milestone was achieved in April 2022, at which time the Company incurred a $0.1 million license fees which was recorded as
acquired in-process research and development and included in research and development expenses in the consolidated statement of operations
for the year ended September 30, 2022. No license fees were incurred during the year ended September 30, 2023.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
In
October 2021, the Company entered into a Non-Exclusive License Agreement (the “Brink Agreement”) with Brink Biologics Inc.
(“Brink”), pursuant to which Brink has granted the Company a non-exclusive, non-transferable license and limited right to
sublicense certain materials and related information to develop cell-based assays for batch, quality control, stability, efficacy, potency
or any other type of assay required for production and commercialization of products. During the product development phase, the Company
was obligated to make annual product development license fee payments of approximately $0.1 million. In April 2023, the Brink Agreement
was amended, effective November 2022, to reduce the annual license fee payments to $12,000 for storage. If materials are removed from
storage during the product development phase, the annual product development license fee of approximately $0.1 million will apply. If
a product achieves commercial status, the Company is obligated to make a commercial product license fee payment of approximately $0.1
million per commercial product. The amended agreement has an initial term of one year and will automatically renew for one additional
year unless terminated or converted to a product development license. After the second year, the license will automatically convert to
a full license requiring a product development or a commercial product license fee unless the parties mutually agree to terminate the
agreement. The Company incurred $12,000 and $0.2 million in license fees during the years ended September 30, 2023 and 2022, respectively,
which were recorded as acquired in-process research and development and included in research and development expenses in the consolidated
statements of operations.
In
February 2022, the Company entered into a Biological Materials License Agreement (the “InvivoGen Agreement”) with InvivoGen
SAS (“InvivoGen”), pursuant to which InvivoGen has granted the Company a worldwide, non-exclusive license to use certain
reporter cells for research, development and/or quality control purposes. The InvivoGen Agreement has an initial term of three years
and may be extended for two additional three-year periods upon written notice by the Company and payment of an approximately €0.1
million fee per extension (approximately $0.1 million as of September 30, 2023). The Company incurred a $0.1 million license fee which
was recorded as acquired in-process research and development and included in research and development expenses in the consolidated statement
of operations for the year ended September 30, 2022. No license fees were incurred for the year ended September 30, 2023.
In
March 2022, the Company entered into a Material Transfer and License Agreement (the “ProteoNic Agreement”) with ProteoNic
B.V. (“ProteoNic”), pursuant to which ProteoNic has granted to the Company a non-exclusive, non-transferable, non-sublicensable
(except as provided for in the ProteoNic Agreement) license for certain materials, including plasmids and DNA sequences used to generate
the vectors used in the Company’s cell lines, for the Company’s use in research, development and commercialization of product.
The license will continue until terminated by either party. The Company incurred a $24,600 license fee upon obtaining the license. The
Company is obligated to make contingent milestone payments to ProteoNic totaling up to €1.2 million (approximately $1.3 million
as of September 30, 2023) upon the achievement of certain development and commercialization milestones as outlined in the ProteoNic Agreement.
The Company incurred a $24,600 license fee which was recorded as acquired in-process research and development and included in research
and development expenses in the consolidated statement of operations for the year ended September 30, 2022. No license fees were incurred
during the year ended September 30, 2023.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
Research
and development agreement
In
December 2021, the Company entered into a Research and Development Agreement (the “Navigo Agreement”) with Navigo Proteins
GmbH (“Navigo”), pursuant to which Navigo will perform specified evaluation and development procedures to evaluate certain
materials to determine their commercial potential. Under the terms of the Navigo Agreement, the Company has granted Navigo a royalty-free,
non-exclusive, worldwide, non-sublicensable, non-transferable right and license to use certain technology to perform the evaluation and
development activities, and Navigo has granted the Company (i) an exclusive, worldwide, perpetual, irrevocable, sublicensable, transferable,
royalty-free right and license to research, develop, use, sell, have sold, distribute, import or otherwise commercially exploit certain
materials, and (ii) a non-exclusive, worldwide, perpetual, sublicensable, non-transferable right and license to make or have made such
materials. The Company incurred a $0.1 million technology access fee upon execution of the Navigo Agreement, at which time it was recorded
as acquired in-process research and development and included in research and development expenses in the consolidated statement of operations
for the year ended September 30, 2022. The Company is obligated to make contingent milestone payments to Navigo, as amended in March
2023, totaling up to $1.0 million upon the achievement of certain evaluation and development milestones as outlined in the Navigo Agreement.
Certain evaluation milestones were achieved in 2023, totaling $0.3 million in license fees, which were recorded as acquired in-process
research and development and included as research and development expenses in the consolidated statement of operations for the year ended
September 30, 2023.
Employment
agreements
The
Company has entered into employment contracts with its officers and certain employees that provide for severance and continuation of
benefits in the event of termination of employment either by the Company without cause or by the employee for good reason, both as defined
in the contract. In addition, in the event of termination of employment following a change in control, as defined, either by the Company
without cause or by the employee for good reason, any unvested portion of the employee’s initial stock option grant becomes immediately
vested.
|
| X |
- References
+ Details
| Name: |
us-gaap_CommitmentsAndContingenciesDisclosureAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe entire disclosure for commitments and contingencies. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 440
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482648/440-10-50-4
Reference 2: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 450
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//450/tableOfContent
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 954
-SubTopic 440
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480327/954-440-50-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 440
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482648/440-10-50-4
Reference 5: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 440
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//440/tableOfContent
| Name: |
us-gaap_CommitmentsAndContingenciesDisclosureTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Collaboration Revenue
|
12 Months Ended |
Sep. 30, 2023 |
|---|
| Organization, Consolidation and Presentation of Financial Statements [Abstract] |
|
| Collaboration Revenue |
6.
Collaboration Revenue
Under
the New Life Agreement, the Company granted New Life an exclusive license (with the right to sublicense) to develop and commercialize
pharmaceutical preparations containing a specific recombinant human IL-6, SON-080 (the “Compound”) (such preparations, the
“Products”) for the prevention, treatment or palliation of diabetic peripheral neuropathy in humans (the “DPN Field”)
in Malaysia, Singapore, Indonesia, Thailand, Philippines, Vietnam, Brunei, Myanmar, Lao PDR and Cambodia (the “Exclusive Territory”).
New Life had the option to expand (1) the field of the exclusive license to include the prevention, treatment or palliation of chemotherapy-induced
peripheral neuropathy in humans (the “CIPN Field”), which option was non-exclusive and expired on December 31, 2021; and/or
(2) the territorial scope of the license to include the People’s Republic of China, Hong Kong and/or India, which option was exclusive
and expired on December 31, 2021.
The
Company will retain all rights to manufacture Compounds and Products anywhere in the world. The Company and New Life shall enter into
a follow-on supply agreement pursuant to which the Company shall supply to New Life Products for development and commercialization thereof
in the DPN Field in the Exclusive Territory on terms to be negotiated by the parties. The Company will also assist in transferring certain
preclinical and clinical development know-how that is instrumental in New Life’s ability to benefit from the license.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
New
Life will bear the cost of, and be responsible for, among other things, conducting clinical studies and additional non-clinical studies
and other developmental and regulatory activities for and commercializing Products in the DPN Field in the Exclusive Territory.
New
Life paid the Company a $0.5 million non-refundable upfront cash payment in August 2020 upon executing a letter of intent to negotiate
a license agreement and a $0.5 million non-refundable upfront cash payment in June 2021 in connection with the execution of the New Life
Agreement. New Life is also obligated to pay a non-refundable deferred license fee of an additional $1.0 million at the time of the satisfaction
of certain milestones, as well as potential additional milestone payments to the Company of up to $19.0 million subject to the achievement
of certain development and commercialization milestones. In addition, during the Royalty Term (as defined below), New Life is obligated
to pay the Company tiered double-digit royalties ranging from 12% to 30% based on annual net sales of Products in the Exclusive Territory.
The “Royalty Term” means, on a Product-by-Product and a country-by-country basis in the Exclusive Territory, the period commencing
on the date of the first commercial sale (subject to certain conditions) of such Product in such country in the Exclusive Territory and
continuing until New Life ceases commercialization of such Product in the DIPN Field.
The
New Life Agreement will remain in effect on a Product-by-Product, country-by-country basis and will expire upon the expiration of the
Royalty Term for the last-to-expire Product in the last-to-expire country, subject to (i) each party’s early termination rights
including for material breach or insolvency or bankruptcy of the other party and (ii) the Company’s Buy Back Right and New Life’s
Give Back Right (as defined below).
In
addition, New Life granted to the Company an exclusive option to buy back the rights granted by the Company to New Life and the Company
granted New Life the right to give back the rights with respect to Products in the DPN Field in one or more countries in the Exclusive
Territory on terms to be agreed upon, which options will expire upon the initiation of a Phase III Trial for the applicable Product.
Revenue
recognition
The
Company first assessed the New Life Agreement under ASC 808, Collaborative Arrangements (“ASC 808”), to determine
whether the New Life Agreement or units of accounts within the New Life Agreement represent a collaborative arrangement based on the
risks and rewards and activities of the parties. The Company applied relevant guidance from ASC 606, Revenue from Contracts with Customers
(“ASC 606”), to evaluate the appropriate accounting for the collaborative arrangement with New Life. In accordance with
this guidance, the Company identified the following obligations under the arrangement: (i) License to develop, market, import, use and
commercialize the Product in the Field in the Exclusive Territory (the “License”); and (ii) transfer of know-how and clinical
development and regulatory activities (“R&D Activities”). The options to expand the CIPN Field and territory as well
as the future supply agreement represent optional purchases, which are accounted for as separate contracts. The Company evaluated these
separate contracts and did not identify any material right to be present. The Company determined that License and the R&D services
are not distinct from each other and therefore combined these material promises into a single performance obligation.
The
Company determined the initial transaction price of the single performance obligation to be $1.0 million, as the future development and
commercialization milestones, which represent variable consideration, are subject to constraint at inception. At the end of each subsequent
reporting period, the Company will reevaluate the probability of achievement of the future development and commercialization milestones
subject to constraint and, if necessary, will adjust its estimate of the overall transaction price. Any such adjustments will be recorded
on a cumulative catch-up basis. For the sales-based royalties, the Company will recognize revenue when the related sales occur.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
Collaboration
revenue from the single performance obligation is being recognized over the estimated performance of the R&D services. The Company
recognized $0.1 million and $0.3 million of collaboration revenue for the years ended September 30, 2023 and 2022, respectively.
|
| X |
- DefinitionThe entire disclosure for collaborative arrangements in which the entity is a participant, including a) information about the nature and purpose of such arrangements; b) its rights and obligations thereunder; c) the accounting policy for collaborative arrangements; and d) the income statement classification and amounts attributable to transactions arising from the collaborative arrangement between participants. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (a)
-SubTopic 10
-Topic 808
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479402/808-10-50-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (b)
-SubTopic 10
-Topic 808
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479402/808-10-50-1
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Topic 808
-Publisher FASB
-URI https://asc.fasb.org//808/tableOfContent
| Name: |
us-gaap_CollaborativeArrangementDisclosureTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_OrganizationConsolidationAndPresentationOfFinancialStatementsAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Stockholders’ Deficit
|
12 Months Ended |
Sep. 30, 2023 |
|---|
| Equity [Abstract] |
|
| Stockholders’ Deficit |
7.
Stockholders’ Deficit
2023
events
The
Company entered into an At-the-Market Sales Agreement with BTIG, LLC (“BTIG”) on August 15, 2022 (the “2022 Sales Agreement”).
Pursuant to the 2022 Sales Agreement, the Company could offer and sell, from time to time, through BTIG, as sales agent and/or principal,
shares of its common stock having an aggregate offering price of up to $25.0 million, subject to certain limitations on the amount of
common stock that may be offered and sold by the Company set forth in the 2022 Sales Agreement. Due to the offering limitations applicable
to the Company, the Company filed prospectus supplements for the sale of shares of its common stock for an aggregate offering price of
up to $7.8 million pursuant to the 2022 Sales Agreement. During the year ended September 30, 2023, the Company sold an aggregate of 136,702
shares of common stock pursuant to the 2022 Sales Agreement with BTIG for gross proceeds of $5.7 million and net proceeds of $5.5 million.
There are no registered shares remaining to be sold under the 2022 Sales Agreement.
On
February 10, 2023, the Company closed a public offering of common stock and certain warrants through Chardan Capital Markets, LLC and
EF Hutton, division of Benchmark Investments LLC as underwriters, for gross proceeds of $15.0 million and net proceeds of $13.6 million
through the issuance and sale of 530,222 shares of its common stock and, to certain investors, pre-funded warrants to purchase 101,090
shares of common stock, and accompanying common warrants to purchase up to an aggregate of 1,262,618 shares of its common stock (the
“February Offering”). Each share of common stock and pre-funded warrant to purchase one share of common stock was sold together
with a common warrant to purchase two shares of common stock. The public offering price of each share of common stock and accompanying
common warrant was $23.76 and the public offering price of each pre-funded warrant and accompanying common warrant was $23.7578.
The
common stock warrants are immediately exercisable at a price of $23.76 per share of common stock, expire five years from the date of
issuance and contain an alternative cashless exercise provision whereby, subject to certain conditions, a warrant may be exercised in
a cashless transaction for shares of common stock at the rate of half a share of common stock per full share otherwise issuable upon
a cash exercise. The pre-funded warrants are immediately exercisable at any time, until exercised in full, at a price of $0.0022 per
share of common stock.
In
addition, warrants to purchase 44,190 shares of common stock were issued to the underwriters as compensation for their services related
to the offering. These common stock warrants have an exercise price of $29.70 per share and expire five years from the date of issuance.
On
June 30, 2023, the Company closed a registered direct offering of common stock (and common stock equivalents in lieu thereof) and a concurrent
private placement of certain common stock warrants through Chardan Capital Markets, LLC as placement agent, for gross proceeds of $2.3
million and net proceeds of $1.9 million through the issuance and sale of 166,363 shares of its common stock and, to certain investors,
pre-funded warrants to purchase 60,909 shares of common stock, and accompanying common warrants to purchase up to an aggregate of 227,272
shares of its common stock (the “June Offering”). Each share of common stock and pre-funded warrant to purchase one share
of common stock was sold together with a common warrant to purchase one share of common stock. The public offering price of each share
of common stock and accompanying common warrant was $9.90.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
The
common stock warrants are exercisable beginning December 30, 2023 at a price of $14.8478 per share of common stock, expire three and
half years from the date of issuance and contain an alternative cashless exercise provision. The pre-funded warrants are immediately
exercisable at any time, until exercised in full, at a price of $0.0022 per share of common stock. All of the pre-funded warrants have
been exercised.
In
addition, warrants to purchase 6,818 shares of common stock were issued to the placement agent as compensation for its services related
to the offering. These common stock warrants have an exercise price of $14.8478 per share and expire three and a half years from the
date of issuance.
2022
events
In
August 2022, the Company entered into a securities purchase agreement (the “Preferred SPA”) with several accredited investors
for the issuance and sale of (i) an aggregate of 22,275 shares of its Series 3 Convertible Preferred Stock, stated value $100 per share,
(ii) 225 shares of its Series 4 Convertible Preferred Stock, stated value $100 per share, and (iii) Series 3 warrants to purchase up
to 12,548 shares of its common stock in a private placement for aggregate gross proceeds of $2.3 million, with $0.1 million of issuance
costs for net proceeds of $2.1 million. The shares of Series 3 Convertible Preferred Stock were convertible into an aggregate of 24,850
shares of common stock of the Company and the shares of Series 4 Convertible Preferred Stock were convertible into an aggregate of 251
shares of common stock of the Company, in each case, at a conversion price of $89.628 per share. The Series 3 Warrants have an exercise
price of $89.628 per share, are exercisable commencing six months after issuance, and will expire five years from the issuance date.
The net proceeds from the private placement were allocated to the Series 3 Convertible Preferred Stock, Series 4 Convertible Preferred
Stock and Series 3 warrants based on their relative fair values.
The
shares of the Series 3 and Series 4 Convertible Preferred Stock had no voting rights other than the right to vote with common stockholders,
as a single class, on a proposed amendment to the Company’s Certificate of Incorporation, as amended, to effect a reverse split
of the outstanding shares of common stock. Each share of Series 3 Convertible Preferred Stock was entitled to vote on an as-converted
basis, and each share of Series 4 Convertible Preferred Stock was entitled to 811,688 votes per share, provided that the Series 4 Convertible
Preferred Stock be automatically voted in the same proportions as the shares of common stock and Series 3 Convertible Preferred Stock
on the reverse split proposal. The shares of the Series 3 and Series 4 Convertible Preferred Stock were convertible at the option of
the holder at any time following the effective date of a reverse split of the Company’s outstanding common stock. In addition,
the Company could elect mandatory conversion on the date of the reverse stock split or the Company could force conversion of all or a
part of the Series 3 and Series 4 Convertible Preferred Stock at any time after 120 days following the issuance of the preferred stock,
in either case provided certain equity conditions were met.
The
private placement closed on August 15, 2022, and the stockholders voted to approve a 1-for-14 reverse stock split on September 15, 2022.
The Series 3 and Series 4 Convertible Preferred Stock were converted to 25,101 shares of common stock on September 30, 2022. As of September
30, 2023 and 2022, there were no shares of preferred stock issued and outstanding.
During
the year ended September 30, 2022, the Company sold an aggregate of 30,206 shares of common stock pursuant to the 2022 Sales Agreement
with BTIG for gross proceeds of $2.0 million and net proceeds of $1.9 million.
Also
during the year ended September 30, 2022, the Company issued 1,087 shares of common stock upon the vesting of restricted stock units.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
Common
stock warrants
As
of September 30, 2023, the following equity-classified warrants and related terms were outstanding:
Schedule
of Warrants Outstanding
| | |
Warrants
Outstanding | | |
Exercise
Price | | |
Expiration
Date |
| Common
stock warrants August 2021 | |
| 128,500 | | |
$ | 261.80 | | |
August
24, 2024 |
| Underwriter
warrants August 2021 | |
| 2,287 | | |
$ | 327.25 | | |
August
19, 2024 |
| Chanticleer
warrants | |
| 57 | | |
$ | 18,018.00
-$28,028.00 | | |
April
30, 2027 - December 17, 2028 |
| Series
C warrants | |
| 36,778 | | |
$ | 982.52 | | |
October
16, 2025 |
| Series
3 warrants | |
| 12,548 | | |
$ | 89.628 | | |
August
15, 2027 |
| Common
stock warrants February 2023 | |
| 271,883 | | |
$ | 23.76 | | |
February
10, 2028 |
| Underwriter
warrants February 2023 | |
| 44,190 | | |
$ | 29.70 | | |
February
8, 2028 |
| Common
stock private placement warrants June 2023 | |
| 227,272 | | |
$ | 14.8478 | | |
December
30, 2026 |
| Placement
agent warrants June 2023 | |
| 6,818 | | |
$ | 14.8478 | | |
December
30, 2026 |
| Total | |
| 730,333 | | |
| | | |
|
During
the year ended September 30, 2023, 1,014,872 warrants were net share settled, resulting in the issuance of 519,492 shares of common stock.
During
the year ended September 30, 2023, 137,999 warrants were exercised on a cash basis. The Company received de minimus proceeds in exchange
for the issuance of 137,998 shares of common stock.
During
the year ended September 30, 2023, 332 of private warrants expired.
|
| X |
- References
+ Details
| Name: |
us-gaap_EquityAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe entire disclosure for equity. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 13
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-13
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 13
-Subparagraph (h)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-13
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 14
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-14
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 235
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481062/946-235-50-2
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 235
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481062/946-235-50-2
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 505
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481004/946-505-50-6
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 815
-SubTopic 40
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480237/815-40-50-6
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(e)(1))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 10: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//505/tableOfContent
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 13
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-13
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 13
-Subparagraph (g)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-13
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 13
-Subparagraph (i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-13
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 14
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-14
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 14
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-14
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 16
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-16
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 18
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-18
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 18
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-18
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 18
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-18
| Name: |
us-gaap_StockholdersEquityNoteDisclosureTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Share-Based Compensation
|
12 Months Ended |
Sep. 30, 2023 |
|---|
| Share-Based Payment Arrangement [Abstract] |
|
| Share-Based Compensation |
8.
Share-Based Compensation
In
April 2020, the Company adopted the 2020 Omnibus Equity Incentive Plan (the “Plan”). On January 1, 2023, the total number
of shares authorized under the Plan increased to 14,480. There were 14,480 shares available for issuance under the Plan as of September
30, 2023. The Plan increases the amount of shares issuable under the Plan by four percent of the outstanding shares of common stock at
each January 1, each year. The Plan permits the granting of share-based awards, including stock options, restricted stock units and awards,
stock appreciation rights and other types of awards as deemed appropriate, in each case, in accordance with the terms of the Plan. The
terms of the awards are determined by the Company’s Board of Directors.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
Restricted
stock units and awards
In
July of 2020, 2,115 restricted stock units (“RSUs”) were granted, 50% of which vested on April 2, 2021 and the remaining
50% vested on April 2, 2022. In March of 2021, an additional 152 RSUs were granted, 50% of which vested on March 25, 2022 and the remaining
50% vested on March 25, 2023. In December of 2021, 2,103 RSUs were granted, 100% of which vested on January 1, 2023. In December of 2022,
7,840 RSUs were granted, 100% of which vest on January 1, 2024.
In
January 2023, 5,514 of the RSUs granted in December 2022 were cancelled and subsequently reissued as restricted shares of the Company’s
common stock (“Restricted Stock Awards” or “RSAs”). The RSAs have the same vesting conditions as the original
RSUs issued in December 2022. The Company accounted for this as a stock compensation modification resulting in $38,837 of incremental
expense which will be recognized over the remaining vesting period.
Any
unvested RSUs or RSAs will be forfeited upon termination of services. The fair value of an RSU or RSA is equal to the fair market value
of the Company’s common stock on the date of grant. RSU and RSA expense is amortized straight-line over the vesting period.
The
Company recorded share-based compensation expense associated with the RSUs and RSAs in its accompanying consolidated statements of operations
as follows:
Schedule
of Share-based Compensation Expense
| | |
2023 | | |
2022 | |
| | |
Years
ended September 30, | |
| | |
2023 | | |
2022 | |
| Research
and development | |
$ | 121,265 | | |
$ | 437,921 | |
| General
and administrative | |
| 127,361 | | |
| 438,447 | |
| Share
Based Compensation | |
$ | 248,626 | | |
$ | 876,368 | |
The
following table summarizes RSU activity under the Plan:
Schedule
of Restricted Stock Units Activity
| | |
| | |
Weighted | |
| | |
| | |
Average
Grant | |
| | |
RSU | | |
Date
Fair Value | |
| Unvested
balance at October 1, 2021 | |
| 1,179 | | |
$ | 1,068.32 | |
| Granted | |
| 2,103 | | |
$ | 155.54 | |
| Vested | |
| (1,096 | ) | |
$ | 1,091.20 | |
| Forfeited | |
| (24 | ) | |
$ | 1,118.04 | |
| Unvested
balance at October 1, 2022 | |
| 2,162 | | |
$ | 175.56 | |
| Granted | |
| 7,840 | | |
$ | 21.47 | |
| Vested | |
| (2,162 | ) | |
$ | 12.55 | |
| Forfeited | |
| (5,514 | ) | |
$ | 21.34 | |
| Unvested
balance at September 30, 2023 | |
| 2,326 | | |
$ | 21.78 | |
As
of September 30, 2023, total unrecognized compensation expense relating to unvested RSUs granted was $12,194, which is expected to be
recognized over a weighted-average period of three months.
The
following table summarizes RSA activity under the Plan:
Schedule
of Restricted Stock Awards Activity
| | |
| | |
Weighted | |
| | |
| | |
Average
Grant | |
| | |
RSA | | |
Date
Fair Value | |
| Unvested
balance at October 1, 2022 | |
| — | | |
| — | |
| Granted | |
| 5,514 | | |
$ | 28.27 | |
| Unvested
balance at September 30, 2023 | |
| 5,514 | | |
$ | 28.27 | |
As
of September 30, 2023, total unrecognized compensation expense relating to unvested RSAs granted was $37,812, which is expected to be
recognized over a weighted-average period of three months.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
|
| X |
- DefinitionThe entire disclosure for share-based payment arrangement. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//718/tableOfContent
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (h)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (h)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (l)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_DisclosureOfCompensationRelatedCostsShareBasedPaymentsTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Income Taxes
|
12 Months Ended |
Sep. 30, 2023 |
|---|
| Income Tax Disclosure [Abstract] |
|
| Income Taxes |
9.
Income Taxes
As
of September 30, 2023, the Company had $103.6
million, $69.5
million and $8.9
million of federal, state and foreign net operating losses, respectively. The federal and state net operating losses will begin to
expire in 2030 and the foreign net operating losses begin to expire
in 2027. As of September 30, 2023, the Company has federal and state research and development tax credit carryforwards of
$2.4
million and $0.8
million available to reduce future tax liabilities which will begin to expire in 2035 and 2030, respectively. Realization of the
deferred tax asset is contingent on future taxable income and based upon the level of historical losses, management has concluded
that the deferred tax asset does not meet the more-likely-than-not threshold for realizability. Accordingly, a full valuation
allowance continues to be recorded against the Company’s deferred tax assets as of September 30, 2023 and 2022. The valuation
allowance increased $5.8
million during the year ended September 30, 2023 and $8.3
million during the year ended September 30, 2022.
Due
to the change in ownership provisions of the Internal Revenue Code, the availability of the Company’s net operating loss carryforwards
may be subject to annual limitations, against taxable income in future periods, which could substantially limit the eventual utilization
of such carryforwards. The Company has not analyzed the historical or potential impact of its equity financings on beneficial ownership
and therefore no determination has been made whether the net operating loss carryforwards are subject to any Internal Revenue Code Section
382 limitation. To the extent there is a limitation, there would be a reduction in the deferred tax assets with an offsetting reduction
in the valuation allowance.
When
uncertain tax positions exist, the Company recognizes the tax benefit of tax positions to the extent that the benefit will more likely-than-not
be realized. The determination as to whether the tax benefit will more-likely-than-not be realized is based upon the technical merits
of the tax position as well as consideration of the available facts and circumstances. The Company recognizes interest and penalties
accrued on any unrecognized tax benefits within the provision for income taxes in its consolidated statements of operations. No unrecognized
tax benefits have been recorded.
The
tax effects of the temporary differences that gave rise to deferred taxes were as follows:
Schedule of Deferred Tax Assets and Liabilities
| | |
2023 | | |
2022 | |
| | |
September
30, | |
| | |
2023 | | |
2022 | |
| Deferred
tax assets: | |
| | | |
| | |
| Net
operating loss carryforwards | |
$ | 27,996,751 | | |
$ | 25,858,311 | |
| Research
and development credit carryforwards | |
| 3,106,675 | | |
| 2,151,942 | |
| Amortization | |
| 4,692,227 | | |
| 2,897,388 | |
| Share-based
compensation | |
| 226 | | |
| 66,832 | |
| Operating
lease liability | |
| 57,319 | | |
| 71,748 | |
| Accrued
expenses and other | |
| 546,612 | | |
| 314,210 | |
| Section
163(j) disallowed interest expense | |
| 763,172 | | |
| — | |
| Property
and equipment | |
| — | | |
| 4,790 | |
| Gross
deferred tax assets | |
| 37,162,982 | | |
| 31,365,221 | |
| Less:
valuation allowance | |
| (37,100,582 | ) | |
| (31,293,092 | ) |
| Deferred tax assets | |
| 62,400 | | |
| 72,129 | |
| Deferred
tax liabilities: | |
| | | |
| | |
| Property
and equipment | |
| (7,954 | ) | |
| — | |
| Operating
lease right-of-use asset | |
| (54,446 | ) | |
| (72,129 | ) |
| Net
deferred tax assets | |
$ | — | | |
$ | — | |
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
The
Company recorded no income tax expense or benefit for the years ended September 30, 2023 and 2022. A reconciliation of income tax (expense)
benefit at the statutory federal income tax rate and income taxes as reflected in the consolidated financial statements is as follows:
Schedule of Effective Income Tax Rate Reconciliation
| | |
2023 | | |
2022 | |
| | |
Years
ended September 30, | |
| | |
2023 | | |
2022 | |
| U.S.
federal statutory rate | |
| (21.0 | )% | |
| (21.0 | )% |
| State
taxes, net of federal benefit | |
| (7.1 | ) | |
| (6.0 | ) |
| Change
in valuation allowance | |
| 30.8 | | |
| 28.1 | |
| Research
and development credit | |
| (5.1 | ) | |
| (3.0 | ) |
| Permanent
differences | |
| (1.6 | ) | |
| 0.7 | |
| Foreign
tax rate differential | |
| 0.3 | | |
| 0.8 | |
| State NOLs | |
| 3.7 | | |
| — | |
| Other | |
| 0.0 | | |
| 0.4 | |
| Effective
income tax rate | |
| — | % | |
| — | % |
In
August 2022, the U.S. enacted the Inflation Reduction Act of 2022 (“IRA”). The IRA contains a number of tax-related provisions
that will be effective for tax years beginning after December 31, 2022, including a corporate alternative minimum tax of 15% on certain
large corporations and an excise tax of 1% on corporate stock repurchases. The Company is currently evaluating the various provisions
of the IRA and does not anticipate a material impact on its consolidated financial statements.
|
| X |
- DefinitionThe entire disclosure for income taxes. Disclosures may include net deferred tax liability or asset recognized in an enterprise's statement of financial position, net change during the year in the total valuation allowance, approximate tax effect of each type of temporary difference and carryforward that gives rise to a significant portion of deferred tax liabilities and deferred tax assets, utilization of a tax carryback, and tax uncertainties information. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 13
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480990/946-20-50-13
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.4-08(h)(2))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480678/235-10-S99-1
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//740/tableOfContent
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 14
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-14
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 21
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-21
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 270
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482526/740-270-50-1
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 17
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-17
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SAB TOPIC 6.I.5.Q1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479360/740-10-S99-1
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SAB Topic 11.C)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479360/740-10-S99-2
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 30
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482603/740-30-50-2
| Name: |
us-gaap_IncomeTaxDisclosureTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Subsequent Events
|
12 Months Ended |
Sep. 30, 2023 |
|---|
| Subsequent Events [Abstract] |
|
| Subsequent Events |
10.
Subsequent Events
The
Company has evaluated subsequent events from the balance sheet date through December 14, 2023, the date at which the consolidated financial statements
were available to be issued. Refer to Note 1 for information regarding the October Offering.
In
December 2023, the Company received preliminary approval of its application to sell up to $4.8 million of its New Jersey state net operating
losses through the Technology Business Tax Certificate Transfer Program (the “Program”). As outlined in the Program, although
the sale has been approved and a buyer identified, the Company must still execute an arrangement with such buyer to consummate a sale
of the state net operating losses.
|
| X |
- References
+ Details
| Name: |
us-gaap_SubsequentEventsAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe entire disclosure for significant events or transactions that occurred after the balance sheet date through the date the financial statements were issued or the date the financial statements were available to be issued. Examples include: the sale of a capital stock issue, purchase of a business, settlement of litigation, catastrophic loss, significant foreign exchange rate changes, loans to insiders or affiliates, and transactions not in the ordinary course of business. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 855
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//855/tableOfContent
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 855
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483399/855-10-50-2
| Name: |
us-gaap_SubsequentEventsTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Summary of Significant Accounting Policies (Policies)
|
12 Months Ended |
Sep. 30, 2023 |
|---|
| Accounting Policies [Abstract] |
|
| Basis of presentation |
a.
Basis of presentation
The
accompanying consolidated financial statements have been prepared in conformity with U.S. generally accepted accounting principles (“U.S.
GAAP”). Any reference in these notes to applicable guidance is meant to refer to U.S. GAAP as found in the Accounting Standards
Codification (“ASC”) and Accounting Standards Updates (“ASU”) promulgated by the Financial Accounting Standards
Board (“FASB”).
|
| Consolidation |
b.
Consolidation
The
consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries. All intercompany accounts and
transactions have been eliminated in consolidation.
|
| Use of estimates |
c.
Use of estimates
The
preparation of the consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions
that affect the amounts reported in the consolidated financial statements and accompanying notes. Significant estimates and assumptions
reflected in these consolidated financial statements include the accrual of research and development expenses. Estimates and assumptions
are periodically reviewed in-light of changes in circumstances, facts and experience. Changes in estimates are recorded in the period
in which they become known. Actual results could differ from management’s estimates.
|
| Reclassifications |
d.
Reclassifications
Certain
amounts from the prior period have been reclassified to conform with the current period presentation.
|
| Segment information |
e.
Segment information
Operating
segments are defined as components of an enterprise about which separate discrete information is available for evaluation by the chief
operating decision maker, or decision-making group, in deciding how to allocate resources and assess performance. The Company views its
operations and manages its business in one segment.
|
| Fair value of financial instruments |
f.
Fair value of financial instruments
Management
believes that the carrying amounts of the Company’s financial instruments, including accounts payable, approximate fair value due
to the short-term nature of those instruments.
|
| Property and equipment |
g.
Property and equipment
Property
and equipment are recorded at cost and depreciated using the straight-line method over the estimated useful lives of the assets. Expenditures
for repairs and maintenance that do not extend the estimated useful life or improve an asset are expensed as incurred. Upon retirement
or sale, the cost and related accumulated depreciation and amortization of assets disposed of are removed from the accounts, and any
resulting gain or loss is included in the consolidated statement of operations.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
|
| Impairment of long-lived assets |
h.
Impairment of long-lived assets
The
Company reviews long-lived assets, such as property and equipment, for impairment whenever events or changes in circumstances indicate
that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison
of the carrying amount of an asset to the undiscounted future cash flows expected to be generated by that asset. If the carrying amount
of an asset exceeds its estimated undiscounted future cash flows, then an impairment charge is recognized for the amount by which the
carrying value of the asset exceeds the estimated fair value of the asset. There were no impairment charges recorded during the fiscal
years ended September 30, 2023 and 2022.
|
| Deferred offering costs |
i.
Deferred offering costs
Legal
and other costs incurred in relation to equity offerings are capitalized as deferred offering costs and charged against the proceeds
from equity offerings when received. If a financing is abandoned, deferred offering costs are expensed. As of September 30, 2023, the
Company had $49,988 in deferred offering costs associated with the October Offering. As of September 30, 2022, the Company had $0.1 million
in deferred offering costs associated with the 2022 Sales Agreement (see Note 7).
|
| Incentive tax receivable |
j.
Incentive tax receivable
Subsidiary
is eligible to participate in an Australian research and development tax incentive program. As part of this program, Subsidiary is eligible
to receive a cash refund from the Australian Taxation Office for a percentage of the research and development costs expended by Subsidiary
in Australia. The
cash refund is available to eligible companies with annual aggregate revenues of less than $20.0 million (Australian) during the reimbursable
period. The Company estimates the amount of cash
refund it expects to receive related to the Australian research and development tax incentive program and records the incentive when
it is probable (i) the Company will comply with relevant conditions of the program and (ii) the incentive will be received. As of both
September 30, 2023 and 2022, the Company’s estimate of the amount of cash refund it expects to receive for eligible spending related
to the Australian research and development tax incentive program was $0.8
million. For each of the years ended September
30, 2023 and 2022, $0.8
million for the expected net cash refund related to the tax
incentive program was included in research and development expenses. In January 2023, the Company received $1.1
million from the Australian government related
to eligible research and development expenses for the year ended September 30, 2022. In April 2023, the Company refunded the Australian
government for $0.2 million
due to new information on the Company’s clinical trial studies.
|
| Collaboration revenue |
k.
Collaboration revenue
Collaboration
arrangements may contain multiple components, which may include (i) licenses; (ii) research and development activities; and (iii) the
manufacturing and supply of certain materials. Payments pursuant to these arrangements may include non-refundable payments, upfront payments,
milestone payments upon the achievement of significant regulatory and development events, sales milestones and royalties on product sales.
The amount of variable consideration is constrained until it is probable that the revenue is not at a significant risk of reversal in
a future period.
In
determining the appropriate amount of revenue to be recognized as the Company fulfills its obligations under a collaboration arrangement,
the Company performs the following steps: (i) identification of the promised goods or services in the contract; (ii) determination of
whether the promised goods or services are performance obligations, including whether they are capable of being distinct; (iii) measurement
of the transaction price, including the constraint on variable consideration; (iv) allocation of the transaction price to the performance
obligations; and (v) recognition of revenue as the Company satisfies each performance obligation.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
The
Company applies significant judgment when evaluating whether contractual obligations represent distinct performance obligations, allocating
transaction price to performance obligations within a contract, determining when performance obligations have been met, and assessing
the recognition of variable consideration. When consideration is received prior to the Company completing its performance obligation
under the terms of a contract, a contract liability is recorded as deferred income. Deferred income expected to be recognized as revenue
within the twelve months following the balance sheet date is classified as a current liability. In May 2021, the Company entered into
a License Agreement (the “New Life Agreement”) with New Life. See Note 6 for further discussion of the New Life Agreement.
|
| Research and development expense |
l.
Research and development expense
Research
and development expenses include all direct and indirect costs associated with the development of the Company’s biopharmaceutical
products. These expenses include personnel costs, consulting fees, and payments to third parties for research, development, and manufacturing
services. These costs are charged to expense as incurred.
At
the end of the reporting period, the Company compares payments made to third-party service providers to the estimated progress toward
completion of the related project, based on the measure of progress as defined in the contract. Factors the Company considers in preparing
the estimates include costs incurred by the service provider, milestones achieved, and other criteria related to the efforts of its service
providers. Such estimates are subject to change as additional information becomes available. Depending on the timing of payment to the
service providers and the progress that the Company estimates has been made as a result of the service provided, the Company will record
a prepaid expense or accrued liability relating to these costs. Upfront milestone payments made to third parties who perform research
and development services on the Company’s behalf are expensed as services are rendered. Contingent development or regulatory milestone
payments are recognized upon the related resolution of such contingencies.
|
| Foreign currency |
m.
Foreign currency
Transaction
gains and losses resulting from exchange rate changes on transactions denominated in currencies other than the U.S. dollar are included
in operations in the period in which the transaction occurs and reported within the foreign exchange loss line item in the consolidated
statements of operations.
|
| Share-based compensation |
n.
Share-based compensation
The
Company measures equity classified share-based awards granted to employees and nonemployees based on the estimated fair value on the
date of grant and recognizes compensation expense of those awards over the requisite service period, which is the vesting period of the
respective award. The Company accounts for forfeitures as they occur. For share-based awards with service-based vesting conditions, the
Company recognizes compensation expense on a straight-line basis over the service period. The Company classifies share-based compensation
expense in its consolidated statements of operations in the same manner in which the award recipient’s payroll costs are classified
or in which the award recipient’s service payments are classified.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
|
| Income taxes |
o.
Income taxes
The
Company uses the asset-and-liability method of accounting for income taxes. Deferred tax assets and liabilities are recognized for the
estimated future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and
liabilities and their respective tax bases, and operating loss and credit carryforwards. Deferred tax assets and liabilities are measured
using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered
or settled. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not
be realized. The Company recognizes the benefit of an uncertain tax position that it has taken or expects to take on its income tax return
if such a position is more likely than not to be sustained.
|
| Reverse stock split |
p.
Reverse stock split
On
August 31, 2023, the Company filed a Certificate of Amendment to its Certificate of Incorporation, as amended, with the Secretary of
State of the State of Delaware, which effected a 1-for
-22 reverse stock split of the Company’s issued and outstanding shares of common stock. As a result of the reverse
stock split, every 22 shares of common stock issued and outstanding was converted into one share of common stock. The reverse stock
split affected all stockholders uniformly and did not alter any stockholder’s percentage interest in the Company’s
equity. No fractional shares were issued in connection with the reverse stock split. Stockholders who would otherwise be entitled to
a fractional share of common stock were instead entitled to receive a proportional cash payment. The reverse stock split did not
change the par value or authorized number of shares of common stock. All common share and per share amounts presented in the
consolidated financial statements and accompanying notes have been retroactively adjusted to reflect the reverse stock
split.
|
| Net loss per share |
q.
Net loss per share
Basic
net loss per share is computed by dividing net loss by the weighted-average number of shares of common stock outstanding during each
period (and potential shares of common stock that are exercisable for little or no consideration).
Diluted
loss per share includes the effect, if any, from the potential exercise or conversion of securities such as common stock warrants and
stock options which would result in the issuance of incremental shares of common stock. For diluted net loss per share, the weighted-average
number of shares of common stock is the same for basic net loss per share due to the fact that when a net loss exists, dilutive securities
are not included in the calculation as the impact is anti-dilutive.
Sonnet
BioTherapeutics Holdings, Inc.
Notes
to Consolidated Financial Statements
The
following potentially dilutive securities have been excluded from the computation of diluted shares of common stock outstanding as they
would be anti-dilutive:
Schedule of Potentially Dilutive Securities
| | |
2023 | | |
2022 | |
| | |
September
30, | |
| | |
2023 | | |
2022 | |
| Common
stock warrants August 2021 | |
| 128,500 | | |
| 128,500 | |
| Underwriter
warrants August 2021 | |
| 2,287 | | |
| 2,287 | |
| Private
warrants | |
| — | | |
| 332 | |
| Chanticleer
warrants | |
| 57 | | |
| 57 | |
| Series
C warrants | |
| 36,778 | | |
| 36,778 | |
| Series
3 warrants | |
| 12,548 | | |
| 12,548 | |
| Unvested
restricted stock units and awards | |
| 2,326 | | |
| 2,162 | |
| Common
stock warrants February 2023 | |
| 271,883 | | |
| — | |
| Underwriter
warrants February 2023 | |
| 44,190 | | |
| — | |
| Common
stock private placement warrants June 2023 | |
| 227,272 | | |
| — | |
| Placement
agent warrants June 2023 | |
| 6,818 | | |
| — | |
| Total
anti-dilutive weighted average shares | |
| 732,659 | | |
| 182,664 | |
|
| Recent accounting pronouncements |
r.
Recent accounting pronouncements
Recently
adopted
In
May 2021, the FASB issued ASU 2021-04, Earnings Per Share (Topic 260), Debt-Modifications and Extinguishments (Subtopic 470-50), Compensation-Stock
Compensation (Topic 718), and Derivatives and Hedging-Contracts in Entity’s Own Equity (Subtopic 815-40): Issuer’s Accounting
for Certain Modifications or Exchanges of Freestanding Equity-Classified Written Call Options. The amendments in ASU 2021-04 provide
guidance to clarify and reduce diversity in an issuer’s accounting for modifications or exchanges of freestanding equity-classified
written call options (for example, warrants) that remain equity classified after modification or exchange. The adoption of ASU 2021-04
on October 1, 2022 did not have any impact on the consolidated financial statements.
In
November 2021, the Financial Accounting Standards Board, or FASB, issued Accounting Standards Update No. 2021-10, Government Assistance
(Topic 832): Disclosure by Business Entities about Government Assistance, or ASU 2021-10, which improves the transparency of government
assistance received by most business entities by requiring the disclosure of: (1) the types of government assistance received; (2) the
accounting for such assistance; and (3) the effect of the assistance on a business entity’s financial statements. This guidance
is effective for financial statements issued for annual periods beginning after 15 December 2021. The adoption of the guidance on October
1, 2022 did not have a material impact on our consolidated financial statements.
|
| X |
- DefinitionIncentive Tax Receivbale [Policy Text Block]
| Name: |
SONN_IncentiveTaxReceivbalePolicyTextBlock |
| Namespace Prefix: |
SONN_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionReverse Stock Split [Policy Text Block]
| Name: |
SONN_ReverseStockSplitPolicyTextBlock |
| Namespace Prefix: |
SONN_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_AccountingPoliciesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for basis of accounting, or basis of presentation, used to prepare the financial statements (for example, US Generally Accepted Accounting Principles, Other Comprehensive Basis of Accounting, IFRS).
| Name: |
us-gaap_BasisOfAccountingPolicyPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for collaborative arrangements. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (c)
-SubTopic 10
-Topic 808
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479402/808-10-50-1
| Name: |
us-gaap_CollaborativeArrangementAccountingPolicy |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy regarding (1) the principles it follows in consolidating or combining the separate financial statements, including the principles followed in determining the inclusion or exclusion of subsidiaries or other entities in the consolidated or combined financial statements and (2) its treatment of interests (for example, common stock, a partnership interest or other means of exerting influence) in other entities, for example consolidation or use of the equity or cost methods of accounting. The accounting policy may also address the accounting treatment for intercompany accounts and transactions, noncontrolling interest, and the income statement treatment in consolidation for issuances of stock by a subsidiary. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 235
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483426/235-10-50-4
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 810
-SubTopic 10
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481203/810-10-50-1
| Name: |
us-gaap_ConsolidationPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for deferral and amortization of significant deferred charges. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(17))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_DeferredChargesPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for computing basic and diluted earnings or loss per share for each class of common stock and participating security. Addresses all significant policy factors, including any antidilutive items that have been excluded from the computation and takes into account stock dividends, splits and reverse splits that occur after the balance sheet date of the latest reporting period but before the issuance of the financial statements. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 260
-SubTopic 10
-Section 50
-Paragraph 1
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 260
-SubTopic 10
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-2
| Name: |
us-gaap_EarningsPerSharePolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for determining the fair value of financial instruments. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 60
-Paragraph 1
-SubTopic 10
-Topic 820
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482053/820-10-60-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-SubTopic 10
-Topic 825
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482907/825-10-50-1
| Name: |
us-gaap_FairValueOfFinancialInstrumentsPolicy |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for (1) transactions denominated in a currency other than the reporting enterprise's functional currency, (2) translating foreign currency financial statements that are incorporated into the financial statements of the reporting enterprise by consolidation, combination, or the equity method of accounting, and (3) remeasurement of the financial statements of a foreign reporting enterprise in a hyperinflationary economy. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//830/tableOfContent
| Name: |
us-gaap_ForeignCurrencyTransactionsAndTranslationsPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for recognizing and measuring the impairment of long-lived assets. An entity also may disclose its accounting policy for long-lived assets to be sold. This policy excludes goodwill and intangible assets. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 360
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SAB Topic 5.CC)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480091/360-10-S99-2
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 05
-Paragraph 4
-SubTopic 10
-Topic 360
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482338/360-10-05-4
| Name: |
us-gaap_ImpairmentOrDisposalOfLongLivedAssetsPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for income taxes, which may include its accounting policies for recognizing and measuring deferred tax assets and liabilities and related valuation allowances, recognizing investment tax credits, operating loss carryforwards, tax credit carryforwards, and other carryforwards, methodologies for determining its effective income tax rate and the characterization of interest and penalties in the financial statements. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-03(h)(1))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479886/946-10-S99-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 17
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-17
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 9
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-9
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 25
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482525/740-10-45-25
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482525/740-10-45-28
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 19
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-19
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482765/220-10-50-1
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 20
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-20
| Name: |
us-gaap_IncomeTaxPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy pertaining to new accounting pronouncements that may impact the entity's financial reporting. Includes, but is not limited to, quantification of the expected or actual impact.
| Name: |
us-gaap_NewAccountingPronouncementsPolicyPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for reclassification affecting comparability of financial statement. Excludes amendment to accounting standards, other change in accounting principle, and correction of error. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 205
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483504/205-10-50-1
| Name: |
us-gaap_PriorPeriodReclassificationAdjustmentDescription |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for long-lived, physical asset used in normal conduct of business and not intended for resale. Includes, but is not limited to, work of art, historical treasure, and similar asset classified as collections. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-SubTopic 10
-Topic 360
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482099/360-10-50-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(8)(a))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-SubTopic 360
-Topic 958
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480321/958-360-50-6
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (d)
-SubTopic 360
-Topic 958
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480321/958-360-50-1
| Name: |
us-gaap_PropertyPlantAndEquipmentPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for costs it has incurred (1) in a planned search or critical investigation aimed at discovery of new knowledge with the hope that such knowledge will be useful in developing a new product or service, a new process or technique, or in bringing about a significant improvement to an existing product or process; or (2) to translate research findings or other knowledge into a plan or design for a new product or process or for a significant improvement to an existing product or process. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 730
-SubTopic 10
-Name Accounting Standards Codification
-Section 05
-Paragraph 1
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483044/730-10-05-1
| Name: |
us-gaap_ResearchAndDevelopmentExpensePolicy |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for segment reporting. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 47
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482785/280-10-55-47
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 29
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-29
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 41
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-41
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 29
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-29
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 29
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-29
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 29
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-29
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 29
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-29
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 29
-Subparagraph (e)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-29
| Name: |
us-gaap_SegmentReportingPolicyPolicyTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for award under share-based payment arrangement. Includes, but is not limited to, methodology and assumption used in measuring cost. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(v)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SAB Topic 14.C.Q3)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479830/718-10-S99-1
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SAB Topic 14.D.1.Q5)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479830/718-10-S99-1
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SAB Topic 14.D.3.Q2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479830/718-10-S99-1
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SAB Topic 14.D.2.Q6)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479830/718-10-S99-1
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-Name Accounting Standards Codification
-Publisher FASB
-URI https://asc.fasb.org//718/tableOfContent
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationOptionAndIncentivePlansPolicy |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionDisclosure of accounting policy for the use of estimates in the preparation of financial statements in conformity with generally accepted accounting principles. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 275
-SubTopic 10
-Section 50
-Paragraph 9
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482861/275-10-50-9
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 275
-SubTopic 10
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482861/275-10-50-4
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (b)
-SubTopic 10
-Topic 275
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482861/275-10-50-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (c)
-SubTopic 10
-Topic 275
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482861/275-10-50-1
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 11
-SubTopic 10
-Topic 275
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482861/275-10-50-11
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 12
-SubTopic 10
-Topic 275
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482861/275-10-50-12
Reference 7: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 275
-SubTopic 10
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482861/275-10-50-8
| Name: |
us-gaap_UseOfEstimates |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Summary of Significant Accounting Policies (Tables)
|
12 Months Ended |
Sep. 30, 2023 |
|---|
| Accounting Policies [Abstract] |
|
| Schedule of Potentially Dilutive Securities |
The
following potentially dilutive securities have been excluded from the computation of diluted shares of common stock outstanding as they
would be anti-dilutive:
Schedule of Potentially Dilutive Securities
| | |
2023 | | |
2022 | |
| | |
September
30, | |
| | |
2023 | | |
2022 | |
| Common
stock warrants August 2021 | |
| 128,500 | | |
| 128,500 | |
| Underwriter
warrants August 2021 | |
| 2,287 | | |
| 2,287 | |
| Private
warrants | |
| — | | |
| 332 | |
| Chanticleer
warrants | |
| 57 | | |
| 57 | |
| Series
C warrants | |
| 36,778 | | |
| 36,778 | |
| Series
3 warrants | |
| 12,548 | | |
| 12,548 | |
| Unvested
restricted stock units and awards | |
| 2,326 | | |
| 2,162 | |
| Common
stock warrants February 2023 | |
| 271,883 | | |
| — | |
| Underwriter
warrants February 2023 | |
| 44,190 | | |
| — | |
| Common
stock private placement warrants June 2023 | |
| 227,272 | | |
| — | |
| Placement
agent warrants June 2023 | |
| 6,818 | | |
| — | |
| Total
anti-dilutive weighted average shares | |
| 732,659 | | |
| 182,664 | |
|
| X |
- References
+ Details
| Name: |
us-gaap_AccountingPoliciesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of securities (including those issuable pursuant to contingent stock agreements) that could potentially dilute basic earnings per share (EPS) in the future that were not included in the computation of diluted EPS because to do so would increase EPS amounts or decrease loss per share amounts for the period presented, by antidilutive securities. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 260
-SubTopic 10
-Section 50
-Paragraph 1
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
| Name: |
us-gaap_ScheduleOfAntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Accrued Expenses and Other Current Liabilities (Tables)
|
12 Months Ended |
Sep. 30, 2023 |
|---|
| Payables and Accruals [Abstract] |
|
| Schedule of Accrued Expenses and Other Current Liabilities |
Accrued
expenses and other current liabilities consisted of the following:
Schedule
of Accrued Expenses and Other Current Liabilities
| | |
2023 | | |
2022 | |
| | |
September
30, | |
| | |
2023 | | |
2022 | |
| Compensation
and benefits | |
$ | 2,091,196 | | |
$ | 1,218,530 | |
| Research
and development | |
| 913,145 | | |
| 1,593,922 | |
| Professional
fees | |
| 224,031 | | |
| 378,890 | |
| Other | |
| 2,550 | | |
| 2,630 | |
| Accrued
expenses and other current liabilities | |
$ | 3,230,922 | | |
$ | 3,193,972 | |
|
| X |
- References
+ Details
| Name: |
us-gaap_PayablesAndAccrualsAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of the components of accrued liabilities.
| Name: |
us-gaap_ScheduleOfAccruedLiabilitiesTableTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Leases (Tables)
|
12 Months Ended |
Sep. 30, 2023 |
|---|
| Leases [Abstract] |
|
| Schedule of Lease Expenses |
The
components of lease expense for the years ended September 30, 2023 and 2022 are as follows:
Schedule
of Lease Expenses
| Lease
expense | |
2023 | | |
2022 | |
| Operating
lease expense | |
$ | 90,837 | | |
$ | 94,828 | |
| Variable
lease expense | |
| 5,978 | | |
| 5,339 | |
| Total
lease cost | |
$ | 96,815 | | |
$ | 100,167 | |
|
| Schedule of Operating Lease Liabilities |
Cash
flow information related to operating leases for the years ended September 30, 2023 and 2022 is as follows:
Schedule of Operating Lease Liabilities
| Cash
paid for amounts included in the measurement of lease liabilities: | |
2023 | | |
2022 | |
| Operating
cash flows from operating leases | |
$ | 79,259 | | |
$ | 98,101 | |
|
| Schedule of Future Minimum Lease Payments |
Future
minimum lease payments under non-cancellable leases at September 30, 2023 are as follows:
Schedule of Future Minimum Lease Payments
| | |
| | |
| Fiscal
year | |
| | |
| 2024 | |
$ | 93,614 | |
| 2025 | |
| 95,487 | |
| 2026 | |
| 48,216 | |
| Total
undiscounted lease payments | |
| 237,317 | |
| Less:
imputed interest | |
| (33,406 | ) |
| Total
lease liabilities | |
$ | 203,911 | |
|
| X |
- DefinitionSchedule Of Operating Leases Of Lessee [Table Text Block]
| Name: |
SONN_ScheduleOfOperatingLeasesOfLesseeTableTextBlock |
| Namespace Prefix: |
SONN_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of lessee's lease cost. Includes, but is not limited to, interest expense for finance lease, amortization of right-of-use asset for finance lease, operating lease cost, short-term lease cost, variable lease cost and sublease income. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-4
| Name: |
us-gaap_LeaseCostTableTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_LeasesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of undiscounted cash flows of lessee's operating lease liability. Includes, but is not limited to, reconciliation of undiscounted cash flows to operating lease liability recognized in statement of financial position. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-6
| Name: |
us-gaap_LesseeOperatingLeaseLiabilityMaturityTableTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Stockholders’ Deficit (Tables)
|
12 Months Ended |
Sep. 30, 2023 |
|---|
| Equity [Abstract] |
|
| Schedule of Warrants Outstanding |
As
of September 30, 2023, the following equity-classified warrants and related terms were outstanding:
Schedule
of Warrants Outstanding
| | |
Warrants
Outstanding | | |
Exercise
Price | | |
Expiration
Date |
| Common
stock warrants August 2021 | |
| 128,500 | | |
$ | 261.80 | | |
August
24, 2024 |
| Underwriter
warrants August 2021 | |
| 2,287 | | |
$ | 327.25 | | |
August
19, 2024 |
| Chanticleer
warrants | |
| 57 | | |
$ | 18,018.00
-$28,028.00 | | |
April
30, 2027 - December 17, 2028 |
| Series
C warrants | |
| 36,778 | | |
$ | 982.52 | | |
October
16, 2025 |
| Series
3 warrants | |
| 12,548 | | |
$ | 89.628 | | |
August
15, 2027 |
| Common
stock warrants February 2023 | |
| 271,883 | | |
$ | 23.76 | | |
February
10, 2028 |
| Underwriter
warrants February 2023 | |
| 44,190 | | |
$ | 29.70 | | |
February
8, 2028 |
| Common
stock private placement warrants June 2023 | |
| 227,272 | | |
$ | 14.8478 | | |
December
30, 2026 |
| Placement
agent warrants June 2023 | |
| 6,818 | | |
$ | 14.8478 | | |
December
30, 2026 |
| Total | |
| 730,333 | | |
| | | |
|
|
| X |
- References
+ Details
| Name: |
us-gaap_EquityAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of warrants or rights issued. Warrants and rights outstanding are derivative securities that give the holder the right to purchase securities (usually equity) from the issuer at a specific price within a certain time frame. Warrants are often included in a new debt issue to entice investors by a higher return potential. The main difference between warrants and call options is that warrants are issued and guaranteed by the company, whereas options are exchange instruments and are not issued by the company. Also, the lifetime of a warrant is often measured in years, while the lifetime of a typical option is measured in months. Disclose the title of issue of securities called for by warrants and rights outstanding, the aggregate amount of securities called for by warrants and rights outstanding, the date from which the warrants or rights are exercisable, and the price at which the warrant or right is exercisable. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 718
-SubTopic 10
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-1
| Name: |
us-gaap_ScheduleOfStockholdersEquityNoteWarrantsOrRightsTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Share-Based Compensation (Tables)
|
12 Months Ended |
Sep. 30, 2023 |
|---|
| Share-Based Payment Arrangement [Abstract] |
|
| Schedule of Share-based Compensation Expense |
The
Company recorded share-based compensation expense associated with the RSUs and RSAs in its accompanying consolidated statements of operations
as follows:
Schedule
of Share-based Compensation Expense
| | |
2023 | | |
2022 | |
| | |
Years
ended September 30, | |
| | |
2023 | | |
2022 | |
| Research
and development | |
$ | 121,265 | | |
$ | 437,921 | |
| General
and administrative | |
| 127,361 | | |
| 438,447 | |
| Share
Based Compensation | |
$ | 248,626 | | |
$ | 876,368 | |
|
| Schedule of Restricted Stock Units Activity |
The
following table summarizes RSU activity under the Plan:
Schedule
of Restricted Stock Units Activity
| | |
| | |
Weighted | |
| | |
| | |
Average
Grant | |
| | |
RSU | | |
Date
Fair Value | |
| Unvested
balance at October 1, 2021 | |
| 1,179 | | |
$ | 1,068.32 | |
| Granted | |
| 2,103 | | |
$ | 155.54 | |
| Vested | |
| (1,096 | ) | |
$ | 1,091.20 | |
| Forfeited | |
| (24 | ) | |
$ | 1,118.04 | |
| Unvested
balance at October 1, 2022 | |
| 2,162 | | |
$ | 175.56 | |
| Granted | |
| 7,840 | | |
$ | 21.47 | |
| Vested | |
| (2,162 | ) | |
$ | 12.55 | |
| Forfeited | |
| (5,514 | ) | |
$ | 21.34 | |
| Unvested
balance at September 30, 2023 | |
| 2,326 | | |
$ | 21.78 | |
|
| Schedule of Restricted Stock Awards Activity |
The
following table summarizes RSA activity under the Plan:
Schedule
of Restricted Stock Awards Activity
| | |
| | |
Weighted | |
| | |
| | |
Average
Grant | |
| | |
RSA | | |
Date
Fair Value | |
| Unvested
balance at October 1, 2022 | |
| — | | |
| — | |
| Granted | |
| 5,514 | | |
$ | 28.27 | |
| Unvested
balance at September 30, 2023 | |
| 5,514 | | |
$ | 28.27 | |
|
| X |
- DefinitionSchedule Of Nonvested Restricted Stock Awards Activity [Table Text Block]
| Name: |
SONN_ScheduleOfNonvestedRestrictedStockAwardsActivityTableTextBlock |
| Namespace Prefix: |
SONN_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of the changes in outstanding nonvested restricted stock units. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 718
-SubTopic 10
-Section 50
-Paragraph 2
-Subparagraph (c)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ScheduleOfNonvestedRestrictedStockUnitsActivityTableTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of activity for award under share-based payment arrangement. Includes, but is not limited to, outstanding award at beginning and end of year, granted, exercised, forfeited, and weighted-average grant date fair value. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 718
-SubTopic 10
-Section 50
-Paragraph 2
-Subparagraph (c)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (d)
-SubTopic 10
-Topic 718
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (e)
-SubTopic 10
-Topic 718
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ScheduleOfShareBasedCompensationActivityTableTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Income Taxes (Tables)
|
12 Months Ended |
Sep. 30, 2023 |
|---|
| Income Tax Disclosure [Abstract] |
|
| Schedule of Deferred Tax Assets and Liabilities |
The
tax effects of the temporary differences that gave rise to deferred taxes were as follows:
Schedule of Deferred Tax Assets and Liabilities
| | |
2023 | | |
2022 | |
| | |
September
30, | |
| | |
2023 | | |
2022 | |
| Deferred
tax assets: | |
| | | |
| | |
| Net
operating loss carryforwards | |
$ | 27,996,751 | | |
$ | 25,858,311 | |
| Research
and development credit carryforwards | |
| 3,106,675 | | |
| 2,151,942 | |
| Amortization | |
| 4,692,227 | | |
| 2,897,388 | |
| Share-based
compensation | |
| 226 | | |
| 66,832 | |
| Operating
lease liability | |
| 57,319 | | |
| 71,748 | |
| Accrued
expenses and other | |
| 546,612 | | |
| 314,210 | |
| Section
163(j) disallowed interest expense | |
| 763,172 | | |
| — | |
| Property
and equipment | |
| — | | |
| 4,790 | |
| Gross
deferred tax assets | |
| 37,162,982 | | |
| 31,365,221 | |
| Less:
valuation allowance | |
| (37,100,582 | ) | |
| (31,293,092 | ) |
| Deferred tax assets | |
| 62,400 | | |
| 72,129 | |
| Deferred
tax liabilities: | |
| | | |
| | |
| Property
and equipment | |
| (7,954 | ) | |
| — | |
| Operating
lease right-of-use asset | |
| (54,446 | ) | |
| (72,129 | ) |
| Net
deferred tax assets | |
$ | — | | |
$ | — | |
|
| Schedule of Effective Income Tax Rate Reconciliation |
The
Company recorded no income tax expense or benefit for the years ended September 30, 2023 and 2022. A reconciliation of income tax (expense)
benefit at the statutory federal income tax rate and income taxes as reflected in the consolidated financial statements is as follows:
Schedule of Effective Income Tax Rate Reconciliation
| | |
2023 | | |
2022 | |
| | |
Years
ended September 30, | |
| | |
2023 | | |
2022 | |
| U.S.
federal statutory rate | |
| (21.0 | )% | |
| (21.0 | )% |
| State
taxes, net of federal benefit | |
| (7.1 | ) | |
| (6.0 | ) |
| Change
in valuation allowance | |
| 30.8 | | |
| 28.1 | |
| Research
and development credit | |
| (5.1 | ) | |
| (3.0 | ) |
| Permanent
differences | |
| (1.6 | ) | |
| 0.7 | |
| Foreign
tax rate differential | |
| 0.3 | | |
| 0.8 | |
| State NOLs | |
| 3.7 | | |
| — | |
| Other | |
| 0.0 | | |
| 0.4 | |
| Effective
income tax rate | |
| — | % | |
| — | % |
|
| X |
- DefinitionTabular disclosure of the components of net deferred tax asset or liability recognized in an entity's statement of financial position, including the following: the total of all deferred tax liabilities, the total of all deferred tax assets, the total valuation allowance recognized for deferred tax assets. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Paragraph 2
-Section 50
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-2
| Name: |
us-gaap_ScheduleOfDeferredTaxAssetsAndLiabilitiesTableTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTabular disclosure of the reconciliation using percentage or dollar amounts of the reported amount of income tax expense attributable to continuing operations for the year to the amount of income tax expense that would result from applying domestic federal statutory tax rates to pretax income from continuing operations. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Paragraph 12
-Section 50
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-12
| Name: |
us-gaap_ScheduleOfEffectiveIncomeTaxRateReconciliationTableTextBlock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:textBlockItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
Organization and Description of Business (Details Narrative) - USD ($)
|
|
9 Months Ended |
12 Months Ended |
Oct. 26, 2023 |
Sep. 30, 2023 |
Sep. 30, 2023 |
Sep. 30, 2022 |
| Subsidiary, Sale of Stock [Line Items] |
|
|
|
|
| Cash |
|
$ 2,300,000
|
$ 2,300,000
|
|
| Proceeds from sale of common stock |
|
|
21,006,371
|
$ 1,841,918
|
| Cash refund expected to be received |
|
800,000
|
800,000
|
$ 800,000
|
| Net operating losses |
|
$ 4,800,000
|
4,800,000
|
|
| October Offering [Member] | Underwriters [Member] |
|
|
|
|
| Subsidiary, Sale of Stock [Line Items] |
|
|
|
|
| Proceeds from sale of common stock |
$ 4,100,000
|
|
|
|
| Purchase of warrants |
85,312
|
|
|
|
| Exercisable price |
$ 2.00
|
|
|
|
| October Offering [Member] | Underwriters [Member] | Common Stock [Member] |
|
|
|
|
| Subsidiary, Sale of Stock [Line Items] |
|
|
|
|
| Sale of common stock, shares |
1,306,250
|
|
|
|
| Exercisable price |
$ 0.0001
|
|
|
|
| October Offering [Member] | Underwriters [Member] | Prefunded Warrant [Member] |
|
|
|
|
| Subsidiary, Sale of Stock [Line Items] |
|
|
|
|
| Purchase of warrants |
1,537,500
|
|
|
|
| Exercisable price |
$ 1.5999
|
|
|
|
| October Offering [Member] | Underwriters [Member] | Common Warrant [Member] |
|
|
|
|
| Subsidiary, Sale of Stock [Line Items] |
|
|
|
|
| Purchase of warrants |
5,687,500
|
|
|
|
| Exercisable price |
$ 1.60
|
|
|
|
| October Offering [Member] | February 2024 [Member] |
|
|
|
|
| Subsidiary, Sale of Stock [Line Items] |
|
|
|
|
| Proceeds from sale of common stock |
|
|
$ 4,100,000
|
|
| X |
- DefinitionCash refund expected to be received.
| Name: |
SONN_CashRefundExpectedToBeReceived |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of currency on hand as well as demand deposits with banks or financial institutions. Includes other kinds of accounts that have the general characteristics of demand deposits. Excludes cash and cash equivalents within disposal group and discontinued operation. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 852
-SubTopic 10
-Name Accounting Standards Codification
-Section 55
-Paragraph 10
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481372/852-10-55-10
Reference 2: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 12
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-12
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 944
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.7-03(a)(2))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479440/944-210-S99-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(4))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section 45
-Paragraph 21
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480555/946-210-45-21
Reference 6: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(1))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 20
-SubTopic 210
-Topic 946
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480555/946-210-45-20
| Name: |
us-gaap_Cash |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionExercise price per share or per unit of warrants or rights outstanding. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-3
| Name: |
us-gaap_ClassOfWarrantOrRightExercisePriceOfWarrantsOrRights1 |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionNumber of securities into which the class of warrant or right may be converted. For example, but not limited to, 500,000 warrants may be converted into 1,000,000 shares. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-3
| Name: |
us-gaap_ClassOfWarrantOrRightNumberOfSecuritiesCalledByWarrantsOrRights |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of operating loss carryforward, before tax effects, available to reduce future taxable income under enacted tax laws. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 740
-SubTopic 10
-Section 50
-Paragraph 3
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-3
| Name: |
us-gaap_OperatingLossCarryforwards |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionThe cash inflow from the additional capital contribution to the entity. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 14
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-14
| Name: |
us-gaap_ProceedsFromIssuanceOfCommonStock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe number of shares issued or sold by the subsidiary or equity method investee per stock transaction.
| Name: |
us-gaap_SaleOfStockNumberOfSharesIssuedInTransaction |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table.
| Name: |
us-gaap_SubsidiarySaleOfStockLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_SubsidiarySaleOfStockAxis=SONN_OctoberOfferingMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
srt_TitleOfIndividualAxis=SONN_UnderwritersMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementEquityComponentsAxis=us-gaap_CommonStockMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementEquityComponentsAxis=SONN_PrefundedWarrantMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementEquityComponentsAxis=SONN_CommonWarrantMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_AwardTypeAxis=SONN_FebruaryTwoThousandTwentyFourMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.23.3
Schedule of Potentially Dilutive Securities (Details) - shares
|
12 Months Ended |
Sep. 30, 2023 |
Sep. 30, 2022 |
| Antidilutive Securities Excluded from Computation of Earnings Per Share [Line Items] |
|
|
| Total anti-dilutive weighted average shares |
732,659
|
182,664
|
| Common Stock Warrants August 2021 [Member] |
|
|
| Antidilutive Securities Excluded from Computation of Earnings Per Share [Line Items] |
|
|
| Total anti-dilutive weighted average shares |
128,500
|
128,500
|
| Underwriter Warrants August 2021 [Member] |
|
|
| Antidilutive Securities Excluded from Computation of Earnings Per Share [Line Items] |
|
|
| Total anti-dilutive weighted average shares |
2,287
|
2,287
|
| Private Warrants [Member] |
|
|
| Antidilutive Securities Excluded from Computation of Earnings Per Share [Line Items] |
|
|
| Total anti-dilutive weighted average shares |
|
332
|
| Chanticleer Warrants [Member] |
|
|
| Antidilutive Securities Excluded from Computation of Earnings Per Share [Line Items] |
|
|
| Total anti-dilutive weighted average shares |
57
|
57
|
| Series C Warrants [Member] |
|
|
| Antidilutive Securities Excluded from Computation of Earnings Per Share [Line Items] |
|
|
| Total anti-dilutive weighted average shares |
36,778
|
36,778
|
| Series 3 Warrants [Member] |
|
|
| Antidilutive Securities Excluded from Computation of Earnings Per Share [Line Items] |
|
|
| Total anti-dilutive weighted average shares |
12,548
|
12,548
|
| Unvested Restricted Stock Units and Awards [Member] |
|
|
| Antidilutive Securities Excluded from Computation of Earnings Per Share [Line Items] |
|
|
| Total anti-dilutive weighted average shares |
2,326
|
2,162
|
| Common Stock Warrants February 2023 [Member] |
|
|
| Antidilutive Securities Excluded from Computation of Earnings Per Share [Line Items] |
|
|
| Total anti-dilutive weighted average shares |
271,883
|
|
| Underwriter Warrants February 2023 [Member] |
|
|
| Antidilutive Securities Excluded from Computation of Earnings Per Share [Line Items] |
|
|
| Total anti-dilutive weighted average shares |
44,190
|
|
| Common Stock Private Placement Warrants June 2023 [Member] |
|
|
| Antidilutive Securities Excluded from Computation of Earnings Per Share [Line Items] |
|
|
| Total anti-dilutive weighted average shares |
227,272
|
|
| Placement Agent Warrants June 2023 [Member] |
|
|
| Antidilutive Securities Excluded from Computation of Earnings Per Share [Line Items] |
|
|
| Total anti-dilutive weighted average shares |
6,818
|
|
| X |
- DefinitionSecurities (including those issuable pursuant to contingent stock agreements) that could potentially dilute basic earnings per share (EPS) or earnings per unit (EPU) in the future that were not included in the computation of diluted EPS or EPU because to do so would increase EPS or EPU amounts or decrease loss per share or unit amounts for the period presented. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 260
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482662/260-10-50-1
| Name: |
us-gaap_AntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareAmount |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table.
| Name: |
us-gaap_AntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_AntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareByAntidilutiveSecuritiesAxis=SONN_CommonStockWarrantsAugustTwentyTwentyOneMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_AntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareByAntidilutiveSecuritiesAxis=SONN_UnderwriterWarrantsAugustTwentyTwentyOneMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_AntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareByAntidilutiveSecuritiesAxis=SONN_PrivateWarrantsMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_AntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareByAntidilutiveSecuritiesAxis=SONN_ChanticleerWarrantsMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_AntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareByAntidilutiveSecuritiesAxis=SONN_SeriesCWarrantsMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_AntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareByAntidilutiveSecuritiesAxis=SONN_SeriesThreeWarrantsMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_AntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareByAntidilutiveSecuritiesAxis=SONN_UnvestedRestrictedStockUnitsAndAwardsMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_AntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareByAntidilutiveSecuritiesAxis=SONN_CommonStockWarrantsFebruaryTwentyTwentyThreeMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_AntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareByAntidilutiveSecuritiesAxis=SONN_UnderwriterWarrantsFebruaryTwentyTwentyThreeMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_AntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareByAntidilutiveSecuritiesAxis=SONN_CommonStockPrivatePlacementWarrantsJuneTwentyTwentyThreeMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_AntidilutiveSecuritiesExcludedFromComputationOfEarningsPerShareByAntidilutiveSecuritiesAxis=SONN_PAWarrantsJuneTwentyTwentyThreeMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.23.3
Summary of Significant Accounting Policies (Details Narrative) - USD ($)
|
|
1 Months Ended |
9 Months Ended |
12 Months Ended |
Aug. 31, 2023 |
Apr. 30, 2023 |
Jan. 31, 2023 |
Sep. 30, 2023 |
Sep. 30, 2023 |
Sep. 30, 2022 |
| Subsidiary, Sale of Stock [Line Items] |
|
|
|
|
|
|
| Impairment charges |
|
|
|
|
$ 0
|
$ 0
|
| Refund Received Related to Revenue from Different Year, Year Revenue Recognized |
|
|
|
|
The
cash refund is available to eligible companies with annual aggregate revenues of less than $20.0 million (Australian) during the reimbursable
period.
|
|
| Expected net cash refund |
|
|
|
$ 800,000
|
$ 800,000
|
800,000
|
| Proceeds from Income Tax Refunds |
|
|
$ 1,100,000
|
|
|
|
| Cash paid to government |
|
$ 200,000
|
|
|
|
|
| Stockholders equity reverse stock split |
1-for
-22 reverse stock split
|
|
|
|
|
|
| Research and Development Tax Incentive Program [Member] |
|
|
|
|
|
|
| Subsidiary, Sale of Stock [Line Items] |
|
|
|
|
|
|
| Expected net cash refund |
|
|
|
|
800,000
|
800,000
|
| 2022 Sales Agreement [Member] |
|
|
|
|
|
|
| Subsidiary, Sale of Stock [Line Items] |
|
|
|
|
|
|
| Deferred offering costs |
|
|
|
|
|
$ 100,000
|
| October Offering [Member] |
|
|
|
|
|
|
| Subsidiary, Sale of Stock [Line Items] |
|
|
|
|
|
|
| Deferred offering costs |
|
|
|
$ 49,988
|
$ 49,988
|
|
| X |
- Definition
+ References
+ Details
| Name: |
SONN_CashPaidToGovernment |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionCash refund expected to be received.
| Name: |
SONN_CashRefundExpectedToBeReceived |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionSpecific incremental costs directly attributable to a proposed or actual offering of securities which are deferred at the end of the reporting period. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 340
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SAB Topic 5.A)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480341/340-10-S99-1
| Name: |
us-gaap_DeferredOfferingCosts |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionThe aggregate amount of write-downs for impairments recognized during the period for long-lived assets held for abandonment, exchange or sale. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (b)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 360
-SubTopic 10
-Name Accounting Standards Codification
-Section 45
-Paragraph 15
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482130/360-10-45-15
| Name: |
us-gaap_ImpairmentOfLongLivedAssetsToBeDisposedOf |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionDescription of the reverse stock split arrangement. Also provide the retroactive effect given by the reverse split that occurs after the balance sheet date but before the release of financial statements. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 4
-Subparagraph (SAB Topic 4.C)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-4
| Name: |
us-gaap_StockholdersEquityReverseStockSplit |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table.
| Name: |
us-gaap_SubsidiarySaleOfStockLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_AwardTypeAxis=SONN_ResearchAndDevelopmentTaxIncentiveProgramMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_TypeOfArrangementAxis=SONN_SalesAgreementMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_SubsidiarySaleOfStockAxis=SONN_OctoberOfferingMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.23.3
Schedule of Accrued Expenses and Other Current Liabilities (Details) - USD ($)
|
Sep. 30, 2023 |
Sep. 30, 2022 |
| Payables and Accruals [Abstract] |
|
|
| Compensation and benefits |
$ 2,091,196
|
$ 1,218,530
|
| Research and development |
913,145
|
1,593,922
|
| Professional fees |
224,031
|
378,890
|
| Other |
2,550
|
2,630
|
| Accrued expenses and other current liabilities |
$ 3,230,922
|
$ 3,193,972
|
| X |
- DefinitionAccrued compensation and benefits, current.
| Name: |
SONN_AccruedCompensationAndBenefitsCurrent |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAccrued research and development current.
| Name: |
SONN_AccruedResearchAndDevelopmentCurrent |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of liabilities incurred to vendors for goods and services received, and accrued liabilities classified as other, payable within one year or the normal operating cycle, if longer.
| Name: |
us-gaap_AccountsPayableAndOtherAccruedLiabilitiesCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionCarrying value as of the balance sheet date of obligations incurred through that date and payable for professional fees, such as for legal and accounting services received. Used to reflect the current portion of the liabilities (due within one year or within the normal operating cycle if longer). Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.20)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_AccruedProfessionalFeesCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of expenses incurred but not yet paid classified as other, due within one year or the normal operating cycle, if longer. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.20)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_OtherAccruedLiabilitiesCurrent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- References
+ Details
| Name: |
us-gaap_PayablesAndAccrualsAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
v3.23.3
| X |
- DefinitionAmount of lease cost recognized by lessee for lease contract. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 55
-Paragraph 53
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479589/842-20-55-53
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-4
| Name: |
us-gaap_LeaseCost |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_LeasesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of single lease cost, calculated by allocation of remaining cost of lease over remaining lease term. Includes, but is not limited to, single lease cost, after impairment of right-of-use asset, calculated by amortization of remaining right-of-use asset and accretion of lease liability. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 55
-Paragraph 53
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479589/842-20-55-53
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-4
| Name: |
us-gaap_OperatingLeaseCost |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of variable lease cost, excluded from lease liability, recognized when obligation for payment is incurred for finance and operating leases. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 55
-Paragraph 53
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479589/842-20-55-53
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-4
| Name: |
us-gaap_VariableLeaseCost |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
v3.23.3
| X |
- References
+ Details
| Name: |
us-gaap_LeasesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of cash outflow from operating lease, excluding payments to bring another asset to condition and location necessary for its intended use. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 45
-Paragraph 5
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479041/842-20-45-5
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (g)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-4
| Name: |
us-gaap_OperatingLeasePayments |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
v3.23.3
| X |
- References
+ Details
| Name: |
us-gaap_LeasesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of lessee's undiscounted obligation for lease payment for operating lease. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-6
| Name: |
us-gaap_LesseeOperatingLeaseLiabilityPaymentsDue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of lessee's undiscounted obligation for lease payment for operating lease to be paid in next fiscal year following current fiscal year. Excludes interim and annual periods when interim periods are reported from current statement of financial position date (rolling approach). Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-6
| Name: |
us-gaap_LesseeOperatingLeaseLiabilityPaymentsDueNextTwelveMonths |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of lessee's undiscounted obligation for lease payment for operating lease to be paid in third fiscal year following current fiscal year. Excludes interim and annual periods when interim periods are reported from current statement of financial position date (rolling approach). Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-6
| Name: |
us-gaap_LesseeOperatingLeaseLiabilityPaymentsDueYearThree |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of lessee's undiscounted obligation for lease payment for operating lease to be paid in second fiscal year following current fiscal year. Excludes interim and annual periods when interim periods are reported from current statement of financial position date (rolling approach). Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-6
| Name: |
us-gaap_LesseeOperatingLeaseLiabilityPaymentsDueYearTwo |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of lessee's undiscounted obligation for lease payments in excess of discounted obligation for lease payments for operating lease. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-6
| Name: |
us-gaap_LesseeOperatingLeaseLiabilityUndiscountedExcessAmount |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionPresent value of lessee's discounted obligation for lease payments from operating lease. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 45
-Paragraph 1
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479041/842-20-45-1
| Name: |
us-gaap_OperatingLeaseLiability |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
v3.23.3
| X |
- DefinitionAmendment of operating lease right of use asset and liability.
| Name: |
SONN_AmendmentOfOperatingLeaseRightOfUseAssetAndLiability |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- References
+ Details
| Name: |
us-gaap_LeasesAbstract |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionWeighted average discount rate for operating lease calculated at point in time. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 55
-Paragraph 53
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479589/842-20-55-53
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (g)(4)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-4
| Name: |
us-gaap_OperatingLeaseWeightedAverageDiscountRatePercent |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:percentItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionWeighted average remaining lease term for operating lease, in 'PnYnMnDTnHnMnS' format, for example, 'P1Y5M13D' represents reported fact of one year, five months, and thirteen days. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 55
-Paragraph 53
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479589/842-20-55-53
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 842
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (g)(3)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147478964/842-20-50-4
| Name: |
us-gaap_OperatingLeaseWeightedAverageRemainingLeaseTerm1 |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:durationItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
v3.23.3
Commitments and Contingencies (Details Narrative)
€ in Millions |
1 Months Ended |
12 Months Ended |
|
|
|
|
|
|
Apr. 30, 2022
USD ($)
|
Mar. 31, 2022
USD ($)
|
Sep. 30, 2023
USD ($)
|
Sep. 30, 2022
USD ($)
|
Sep. 30, 2023
EUR (€)
|
Apr. 30, 2023
USD ($)
|
Mar. 31, 2023
USD ($)
|
Jan. 31, 2019
USD ($)
|
Jul. 31, 2012
USD ($)
|
| Loss Contingencies [Line Items] |
|
|
|
|
|
|
|
|
|
| Payment to acquire in process research and development |
|
|
$ 443,250
|
$ 810,227
|
|
|
|
|
|
| Discovery Collaboration Agreements [Member] | XOMA [Member] |
|
|
|
|
|
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
|
|
|
|
|
| Milestone payment |
|
|
|
|
|
|
|
|
$ 3,800,000
|
| Discovery Collaboration Agreements [Member] | XOMA [Member] | In Process Research and Development [Member] |
|
|
|
|
|
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
|
|
|
|
|
| License fee |
$ 500,000
|
|
|
|
|
|
|
|
|
| License fees |
|
|
$ 0
|
|
|
|
|
|
|
| Cellca Agreement [Member] |
|
|
|
|
|
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
|
|
|
|
|
| Description of milestone payments |
|
|
The Company is obligated to make milestone payments to Cellca totaling up to $0.7 million upon the achievement of certain development
and approval milestones if the Buy-Out Option is not exercised.
|
|
|
|
|
|
|
| Cellca Agreement [Member] | Maximum [Member] |
|
|
|
|
|
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
|
|
|
|
|
| Milestone payment |
|
|
|
|
|
|
|
$ 700,000
|
|
| The Cellca Agreement [Member] |
|
|
|
|
|
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
|
|
|
|
|
| Milestone payment |
|
|
$ 600,000
|
|
|
|
|
|
|
| The Cellca Agreement [Member] | Maximum [Member] |
|
|
|
|
|
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
|
|
|
|
|
| Payment for annual license fee obligation |
|
|
600,000
|
|
|
|
|
|
|
| The Cellca Agreement [Member] | Minimum [Member] |
|
|
|
|
|
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
|
|
|
|
|
| Payment for annual license fee obligation |
|
|
100,000
|
|
|
|
|
|
|
| The Cellca Agreement [Member] | In Process Research and Development [Member] |
|
|
|
|
|
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
|
|
|
|
|
| License fees |
|
|
0
|
|
|
|
|
|
|
| Commercial product license fee payment obligation |
$ 100,000
|
|
|
|
|
|
|
|
|
| The Brink Agreement [Member] |
|
|
|
|
|
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
|
|
|
|
|
| Commercial product license fee payment obligation |
|
|
100,000
|
|
|
|
|
|
|
| Payment of annual license fee |
|
|
|
|
|
$ 12,000
|
|
|
|
| The Brink Agreement [Member] | In Process Research and Development [Member] |
|
|
|
|
|
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
|
|
|
|
|
| License fee |
|
|
12,000
|
200,000
|
|
|
|
|
|
| Commercial product license fee payment obligation |
|
|
100,000
|
|
|
|
|
|
|
| InvivoGen Agreement [Member] |
|
|
|
|
|
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
|
|
|
|
|
| License fees |
|
|
0
|
|
|
|
|
|
|
| Payment for annual license fee obligation |
|
|
100,000
|
|
€ 0.1
|
|
|
|
|
| InvivoGen Agreement [Member] | In Process Research and Development [Member] |
|
|
|
|
|
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
|
|
|
|
|
| License fees |
|
|
100,000
|
|
|
|
|
|
|
| ProteoNic Agreement [Member] |
|
|
|
|
|
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
|
|
|
|
|
| Milestone payment |
|
|
1,300,000
|
|
€ 1.2
|
|
|
|
|
| License fees |
|
|
0
|
|
|
|
|
|
|
| ProteoNic Agreement [Member] | In Process Research and Development [Member] |
|
|
|
|
|
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
|
|
|
|
|
| License fees |
|
$ 24,600
|
24,600
|
|
|
|
|
|
|
| Navigo Agreement [Member] | Navigo Proteins GmbH [Member] | Technology Service [Member] |
|
|
|
|
|
|
|
|
|
| Loss Contingencies [Line Items] |
|
|
|
|
|
|
|
|
|
| Milestone payment |
|
|
$ 300,000
|
|
|
|
$ 1,000,000.0
|
|
|
| Payment to acquire in process research and development |
|
|
|
$ 100,000
|
|
|
|
|
|
| X |
- DefinitionCommerical product license fee payment obligation.
| Name: |
SONN_CommericalProductLicenseFeePaymentObligation |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionDescription of milestone payments.
| Name: |
SONN_MilestonePaymentsDescription |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAnnual license fee payment.
| Name: |
SONN_PaymentForAnnualLicenseFeeObligation |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionPayment of annual license fee.
| Name: |
SONN_PaymentOfAnnualLicenseFeeObligation |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 460
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482425/460-10-50-3
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 450
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483076/450-20-50-1
Reference 3: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 450
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483076/450-20-50-4
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 450
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483076/450-20-50-4
Reference 5: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 450
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 9
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483076/450-20-50-9
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 450
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 9
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483076/450-20-50-9
| Name: |
us-gaap_LossContingenciesLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe cash outflows from the purchase of net carrying value allocated to in-process research and development costs and materials acquired in a business combination. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 230
-SubTopic 10
-Section 45
-Paragraph 13
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-13
| Name: |
us-gaap_PaymentsToAcquireInProcessResearchAndDevelopment |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_TypeOfArrangementAxis=SONN_DiscoveryCollaborationAgreementsMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
dei_LegalEntityAxis=SONN_XOMAMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_FiniteLivedIntangibleAssetsByMajorClassAxis=us-gaap_InProcessResearchAndDevelopmentMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_TypeOfArrangementAxis=SONN_CellcaAgreementMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
srt_RangeAxis=srt_MaximumMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_TypeOfArrangementAxis=SONN_TheCellcaAgreementMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
srt_RangeAxis=srt_MinimumMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_TypeOfArrangementAxis=SONN_TheBrinkAgreementMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_TypeOfArrangementAxis=SONN_InvivoGenAgreementMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_TypeOfArrangementAxis=SONN_ProteoNicAgreementMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_TypeOfArrangementAxis=SONN_NavigoAgreementMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
dei_LegalEntityAxis=SONN_NavigoProteinsGmbHMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
srt_ProductOrServiceAxis=us-gaap_TechnologyServiceMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.23.3
Collaboration Revenue (Details Narrative) - USD ($)
|
1 Months Ended |
12 Months Ended |
Jun. 30, 2021 |
Aug. 31, 2020 |
Sep. 30, 2023 |
Sep. 30, 2022 |
| Collaborative Arrangement and Arrangement Other than Collaborative [Line Items] |
|
|
|
|
| Collavorative arrangement right and obligation |
|
|
New Life is also obligated to pay a non-refundable deferred license fee of an additional $1.0 million at the time of the satisfaction
of certain milestones, as well as potential additional milestone payments to the Company of up to $19.0 million subject to the achievement
of certain development and commercialization milestones. In addition, during the Royalty Term (as defined below), New Life is obligated
to pay the Company tiered double-digit royalties ranging from 12% to 30% based on annual net sales of Products in the Exclusive Territory.
The “Royalty Term” means, on a Product-by-Product and a country-by-country basis in the Exclusive Territory, the period commencing
on the date of the first commercial sale (subject to certain conditions) of such Product in such country in the Exclusive Territory and
continuing until New Life ceases commercialization of such Product in the DIPN Field
|
|
| Performance obligation |
|
|
$ 1,000,000.0
|
|
| Collaboration revenue |
|
|
147,805
|
$ 349,943
|
| License Agreement [Member] |
|
|
|
|
| Collaborative Arrangement and Arrangement Other than Collaborative [Line Items] |
|
|
|
|
| Proceeds for negotiate license agreement |
|
$ 500,000
|
|
|
| New Life Agreement [Member] |
|
|
|
|
| Collaborative Arrangement and Arrangement Other than Collaborative [Line Items] |
|
|
|
|
| Proceeds for negotiate license agreement |
$ 500,000
|
|
|
|
| Future milestone payments |
|
|
1,000,000.0
|
|
| Milestone payments |
|
|
$ 19,000,000.0
|
|
| New Life Agreement [Member] | Minimum [Member] |
|
|
|
|
| Collaborative Arrangement and Arrangement Other than Collaborative [Line Items] |
|
|
|
|
| Royalties, percentage |
|
|
12.00%
|
|
| New Life Agreement [Member] | Maximum [Member] |
|
|
|
|
| Collaborative Arrangement and Arrangement Other than Collaborative [Line Items] |
|
|
|
|
| Royalties, percentage |
|
|
30.00%
|
|
| X |
- DefinitionDeferred license fee amount for future milestone payments.
| Name: |
SONN_DeferredLicenseFeeAmountForFutureMilestonePayments |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionProceeds for negotiate license agreement.
| Name: |
SONN_ProceedsforNegotiateLicenseAgreement |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionDescription of rights and obligations under the collaborative arrangements. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 808
-SubTopic 10
-Section 50
-Paragraph 1
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479402/808-10-50-1
| Name: |
us-gaap_CollaborativeArrangementRightsAndObligations |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 808
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479402/808-10-50-1
| Name: |
us-gaap_CollaborativeArrangementsAndNoncollaborativeArrangementTransactionsLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount, excluding tax collected from customer, of revenue from satisfaction of performance obligation by transferring promised good or service to customer. Tax collected from customer is tax assessed by governmental authority that is both imposed on and concurrent with specific revenue-producing transaction, including, but not limited to, sales, use, value added and excise. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 924
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SAB Topic 11.L)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479941/924-10-S99-1
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 606
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 5
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479806/606-10-50-5
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 30
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-30
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 42
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-42
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 40
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-40
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 22
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-22
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 32
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-32
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 280
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 41
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482810/280-10-50-41
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 606
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479806/606-10-50-4
| Name: |
us-gaap_RevenueFromContractWithCustomerExcludingAssessedTax |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_TypeOfArrangementAxis=SONN_LicenseAgreementMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_TypeOfArrangementAxis=SONN_NewLifeAgreementMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
srt_RangeAxis=srt_MinimumMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
srt_RangeAxis=srt_MaximumMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.23.3
Schedule of Warrants Outstanding (Details)
|
Sep. 30, 2023
$ / shares
shares
|
| Common Stock Warrants August 2021 [Member] |
|
| Class of Warrant or Right [Line Items] |
|
| Number of warrants, outstanding | shares |
128,500
|
| Exercise Price | $ / shares |
$ 261.80
|
| Expiration date |
Aug. 24, 2024
|
| Underwriter Warrants August 2021 [Member] |
|
| Class of Warrant or Right [Line Items] |
|
| Number of warrants, outstanding | shares |
2,287
|
| Exercise Price | $ / shares |
$ 327.25
|
| Expiration date |
Aug. 19, 2024
|
| Chanticleer Warrants [Member] |
|
| Class of Warrant or Right [Line Items] |
|
| Number of warrants, outstanding | shares |
57
|
| Chanticleer Warrants [Member] | Minimum [Member] |
|
| Class of Warrant or Right [Line Items] |
|
| Exercise Price | $ / shares |
$ 18,018.00
|
| Expiration date |
Apr. 30, 2027
|
| Chanticleer Warrants [Member] | Maximum [Member] |
|
| Class of Warrant or Right [Line Items] |
|
| Exercise Price | $ / shares |
$ 28,028.00
|
| Expiration date |
Dec. 17, 2028
|
| Series C Warrants [Member] |
|
| Class of Warrant or Right [Line Items] |
|
| Number of warrants, outstanding | shares |
36,778
|
| Exercise Price | $ / shares |
$ 982.52
|
| Expiration date |
Oct. 16, 2025
|
| Series 3 Warrants [Member] |
|
| Class of Warrant or Right [Line Items] |
|
| Number of warrants, outstanding | shares |
12,548
|
| Exercise Price | $ / shares |
$ 89.628
|
| Expiration date |
Aug. 15, 2027
|
| Common Stock Warrants February 2023 [Member] |
|
| Class of Warrant or Right [Line Items] |
|
| Number of warrants, outstanding | shares |
271,883
|
| Exercise Price | $ / shares |
$ 23.76
|
| Expiration date |
Feb. 10, 2028
|
| Underwriter Warrants February 2023 [Member] |
|
| Class of Warrant or Right [Line Items] |
|
| Number of warrants, outstanding | shares |
44,190
|
| Exercise Price | $ / shares |
$ 29.70
|
| Expiration date |
Feb. 08, 2028
|
| Common Stock Private Placement Warrants June 2023 [Member] |
|
| Class of Warrant or Right [Line Items] |
|
| Number of warrants, outstanding | shares |
227,272
|
| Exercise Price | $ / shares |
$ 14.8478
|
| Expiration date |
Dec. 30, 2026
|
| Placement Agent Warrants June 2023 [Member] |
|
| Class of Warrant or Right [Line Items] |
|
| Number of warrants, outstanding | shares |
6,818
|
| Exercise Price | $ / shares |
$ 14.8478
|
| Expiration date |
Dec. 30, 2026
|
| Warrant [Member] |
|
| Class of Warrant or Right [Line Items] |
|
| Number of warrants, outstanding | shares |
730,333
|
| X |
- DefinitionExercise price per share or per unit of warrants or rights outstanding. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-3
| Name: |
us-gaap_ClassOfWarrantOrRightExercisePriceOfWarrantsOrRights1 |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table.
| Name: |
us-gaap_ClassOfWarrantOrRightLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of warrants or rights outstanding.
| Name: |
us-gaap_ClassOfWarrantOrRightOutstanding |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionExpiration date of outstanding warrant and right embodying unconditional obligation requiring redemption by transferring asset at specified or determinable date or upon event certain to occur, in YYYY-MM-DD format. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 820
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (bbb)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482106/820-10-50-2
| Name: |
us-gaap_WarrantsAndRightsOutstandingMaturityDate |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:dateItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- Details
| Name: |
us-gaap_ClassOfWarrantOrRightAxis=SONN_CommonStockWarrantsAugustTwentyTwentyOneMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_ClassOfWarrantOrRightAxis=SONN_UnderwriterWarrantsAugustTwentyTwentyOneMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_ClassOfWarrantOrRightAxis=SONN_ChanticleerWarrantsMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
srt_RangeAxis=srt_MinimumMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
srt_RangeAxis=srt_MaximumMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_ClassOfWarrantOrRightAxis=SONN_SeriesCWarrantsMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_ClassOfWarrantOrRightAxis=SONN_Series3WarrantsMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_ClassOfWarrantOrRightAxis=SONN_CommonStockWarrantsFebruaryTwentyTwentyThreeMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_ClassOfWarrantOrRightAxis=SONN_UnderwriterWarrantFebruaryTwentyTwentyThreeMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_ClassOfWarrantOrRightAxis=SONN_CommonStockPrivatePlacementWarrantsJuneTwentyTwentyThreeMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_ClassOfWarrantOrRightAxis=SONN_PAWarrantsJuneTwentyTwentyThreeMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_ClassOfWarrantOrRightAxis=us-gaap_WarrantMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.23.3
Stockholders’ Deficit (Details Narrative) - USD ($)
|
|
|
|
|
|
|
1 Months Ended |
12 Months Ended |
|
Aug. 31, 2023 |
Jun. 30, 2023 |
Feb. 10, 2023 |
Sep. 30, 2022 |
Sep. 15, 2022 |
Aug. 15, 2022 |
Jun. 30, 2023 |
Aug. 31, 2022 |
Sep. 30, 2023 |
Sep. 30, 2022 |
Dec. 30, 2023 |
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Offering price for common stock |
|
|
|
|
|
|
|
|
$ 20,895,958
|
$ 1,908,693
|
|
| Net proceeds from issuance of common stock |
|
|
|
|
|
|
|
|
$ 21,006,371
|
1,841,918
|
|
| Stockholders' equity, reverse stock split |
1-for
-22 reverse stock split
|
|
|
|
|
|
|
|
|
|
|
| Conversion of convertible securities |
|
|
|
|
|
|
|
|
|
|
|
| Preferred stock, shares issued |
|
|
|
0
|
|
|
|
|
0
|
0
|
|
| Preferred stock, shares outstanding |
|
|
|
0
|
|
|
|
|
0
|
0
|
|
| Warrant expirations |
|
|
|
|
|
|
|
|
332
|
|
|
| Common Stock [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Offering price for common stock |
|
|
|
|
|
|
|
|
$ 83
|
$ 3
|
|
| Sale of common stock, net of issuance costs, shares |
|
|
|
|
|
|
|
|
833,287
|
30,206
|
|
| Number of convertible shares |
|
|
|
|
|
|
|
|
|
25,101
|
|
| Conversion of convertible securities |
|
|
|
|
|
|
|
|
|
$ 3
|
|
| Shares issued upon vesting |
|
|
|
|
|
|
|
|
|
1,087
|
|
| Net share settlement of warrants, shares |
|
|
|
|
|
|
|
|
519,492
|
|
|
| Prefunded Warrant [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Sale of common stock, net of issuance costs, shares |
|
|
|
|
|
|
|
|
137,998
|
|
|
| Warrants exercised |
|
|
|
|
|
|
|
|
137,999
|
|
|
| Common Stock Warrants [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Sale of common stock, net of issuance costs, shares |
|
|
|
|
|
|
|
|
519,492
|
|
|
| Net share settlement of warrants, shares |
|
|
|
|
|
|
|
|
1,014,872
|
|
|
| Chardan Capital Markets LLC [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Sale of common stock, net of issuance costs, shares |
|
166,363
|
|
|
|
|
|
|
|
|
|
| Warrants to purchase shares |
|
227,272
|
|
|
|
|
227,272
|
|
|
|
|
| Exercisable price |
|
$ 9.90
|
|
|
|
|
$ 9.90
|
|
|
|
|
| Net proceeds from issuance of private placement |
|
$ 1,900,000
|
|
|
|
|
$ 2,300,000
|
|
|
|
|
| 2022 Sales Agreement [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Offering price for common stock |
|
|
|
|
|
$ 7,800,000
|
|
|
|
|
|
| Gross proceeds from sale agreement |
|
|
$ 15,000,000.0
|
|
|
|
|
|
|
|
|
| Net proceeds from issuance of common stock |
|
|
$ 13,600,000
|
|
|
|
|
|
|
|
|
| 2022 Sales Agreement [Member] | Underwriter Warrants [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Warrants to purchase shares |
|
|
44,190
|
|
|
|
|
|
6,818
|
|
|
| Exercisable price |
|
|
$ 29.70
|
|
|
|
|
|
|
|
|
| 2022 Sales Agreement [Member] | Underwriter Warrants [Member] | Forecast [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Exercisable price |
|
|
|
|
|
|
|
|
|
|
$ 14.8478
|
| 2022 Sales Agreement [Member] | Common Stock [Member] | Investors [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Sale of common stock, net of issuance costs, shares |
|
|
530,222
|
|
|
|
|
|
|
|
|
| 2022 Sales Agreement [Member] | Prefunded Warrant [Member] | Investors [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Warrants to purchase shares |
|
|
101,090
|
|
|
|
|
|
|
|
|
| Exercisable price |
|
|
$ 0.0022
|
|
|
|
|
|
|
|
|
| Warrants issue price per share |
|
|
$ 23.7578
|
|
|
|
|
|
|
|
|
| 2022 Sales Agreement [Member] | Prefunded Warrant [Member] | Chardan Capital Markets LLC [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Warrants to purchase shares |
|
60,909
|
|
|
|
|
60,909
|
|
|
|
|
| 2022 Sales Agreement [Member] | Common Warrant [Member] | Investors [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Warrants to purchase shares |
|
|
1,262,618
|
|
|
|
|
|
|
|
|
| Exercisable price |
|
|
$ 23.76
|
|
|
|
|
|
|
|
|
| 2022 Sales Agreement [Member] | Common Warrant [Member] | Chardan Capital Markets LLC [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Exercisable price |
|
|
|
|
|
|
|
|
$ 0.0022
|
|
|
| 2022 Sales Agreement [Member] | Common Warrant [Member] | Chardan Capital Markets LLC [Member] | Forecast [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Exercisable price |
|
|
|
|
|
|
|
|
|
|
$ 14.8478
|
| 2022 Sales Agreement [Member] | BTIG, LLC [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Offering price for common stock |
|
|
|
|
|
$ 25,000,000.0
|
|
|
|
|
|
| Sale of stock, number of shares issued |
|
|
|
|
|
|
|
|
136,702
|
|
|
| Gross proceeds from sale agreement |
|
|
|
|
|
|
|
|
$ 5,700,000
|
|
|
| Net proceeds from issuance of common stock |
|
|
|
|
|
|
|
|
$ 5,500,000
|
|
|
| Securities Purchase Agreement [Member] | Accredited Investors [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Stockholders' equity, reverse stock split |
|
|
|
|
1-for-14 reverse stock split
|
|
|
|
|
|
|
| Securities Purchase Agreement [Member] | Accredited Investors [Member] | Series 3 Convertible Preferred Stock [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Sale of stock, number of shares issued |
|
|
|
|
|
|
|
22,275
|
|
|
|
| Preferred stock, par value |
|
|
|
|
|
|
|
$ 100
|
|
|
|
| Conversion of convertible securities |
|
|
|
$ 25,101
|
|
|
|
|
|
|
|
| Securities Purchase Agreement [Member] | Accredited Investors [Member] | Series 4 Convertible Preferred Stock [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Sale of stock, number of shares issued |
|
|
|
|
|
|
|
225
|
|
|
|
| Preferred stock, par value |
|
|
|
|
|
|
|
$ 100
|
|
|
|
| Number of convertible shares |
|
|
|
|
|
|
|
251
|
|
|
|
| Equity instrument conversion price |
|
|
|
|
|
|
|
$ 89.628
|
|
|
|
| Voting rights per share |
|
|
|
|
|
|
|
each share of Series 4 Convertible Preferred Stock was entitled to 811,688 votes per share
|
|
|
|
| Conversion of convertible securities |
|
|
|
$ 25,101
|
|
|
|
|
|
|
|
| Securities Purchase Agreement [Member] | Accredited Investors [Member] | Series 3 Warrants [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Warrants to purchase shares |
|
|
|
|
|
|
|
12,548
|
|
|
|
| Net proceeds from issuance of private placement |
|
|
|
|
|
|
|
$ 2,100,000
|
|
|
|
| Gross proceeds from issuance of private placement |
|
|
|
|
|
|
|
$ 2,300,000
|
|
|
|
| Number of convertible shares |
|
|
|
|
|
|
|
24,850
|
|
|
|
| Equity instrument conversion price |
|
|
|
|
|
|
|
$ 89.628
|
|
|
|
| Securities Purchase Agreement [Member] | Accredited Investors [Member] | Series 3 Warrants [Member] | Private Placement [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Issuance costs |
|
|
|
|
|
|
|
$ 100,000
|
|
|
|
| 2022 Sales Ageement [Member] | BTIG [Member] | Common Stock [Member] |
|
|
|
|
|
|
|
|
|
|
|
| Accumulated Other Comprehensive Income (Loss) [Line Items] |
|
|
|
|
|
|
|
|
|
|
|
| Sale of stock, number of shares issued |
|
|
|
|
|
|
|
|
|
30,206
|
|
| Net proceeds from issuance of common stock |
|
|
|
|
|
|
|
|
|
$ 1,900,000
|
|
| Gross proceeds from issuance of common stock |
|
|
|
|
|
|
|
|
|
$ 2,000,000.0
|
|
| X |
- DefinitionEquity instrument conversion price.
| Name: |
SONN_EquityInstrumentConversionPrice |
| Namespace Prefix: |
SONN_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionGross proceeds from issuance of common stock
| Name: |
SONN_GrossProceedsFromIssuanceOfCommonStock |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionGross proceeds from issuance of private placement.
| Name: |
SONN_GrossProceedsFromIssuanceOfPrivatePlacement |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionStock issued during period value net share settlement of warrants, shares.
| Name: |
SONN_StockIssuedDuringPeriodSharesNetShareSettlementOfWarrants |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionStock issued during period shares net share warrants exercised.
| Name: |
SONN_StockIssuedDuringPeriodSharesNetShareWarrantsExercised |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionWarrants issue price per share.
| Name: |
SONN_WarrantsIssuedPricePerShare |
| Namespace Prefix: |
SONN_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 4
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482765/220-10-50-4
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 220
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 5
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482765/220-10-50-5
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 50
-Paragraph 1
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481674/830-30-50-1
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 45
-Paragraph 17
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481694/830-30-45-17
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 45
-Paragraph 20
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481694/830-30-45-20
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 45
-Paragraph 20
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481694/830-30-45-20
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 45
-Paragraph 20
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481694/830-30-45-20
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 45
-Paragraph 20
-Subparagraph (d)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481694/830-30-45-20
| Name: |
us-gaap_AccumulatedOtherComprehensiveIncomeLossLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionExercise price per share or per unit of warrants or rights outstanding. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-3
| Name: |
us-gaap_ClassOfWarrantOrRightExercisePriceOfWarrantsOrRights1 |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionNumber of securities into which the class of warrant or right may be converted. For example, but not limited to, 500,000 warrants may be converted into 1,000,000 shares. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-3
| Name: |
us-gaap_ClassOfWarrantOrRightNumberOfSecuritiesCalledByWarrantsOrRights |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionFace amount or stated value per share of preferred stock nonredeemable or redeemable solely at the option of the issuer. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 13
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-13
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(28))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_PreferredStockParOrStatedValuePerShare |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionTotal number of nonredeemable preferred shares (or preferred stock redeemable solely at the option of the issuer) issued to shareholders (includes related preferred shares that were issued, repurchased, and remain in the treasury). May be all or portion of the number of preferred shares authorized. Excludes preferred shares that are classified as debt. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 13
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-13
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(28))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_PreferredStockSharesIssued |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionAggregate share number for all nonredeemable preferred stock (or preferred stock redeemable solely at the option of the issuer) held by stockholders. Does not include preferred shares that have been repurchased. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 2
-Subparagraph (SX 210.6-05(4))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-2
Reference 2: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(4)(b))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 210
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.6-04(16)(a))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479617/946-210-S99-1
Reference 4: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(7))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 5: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(28))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_PreferredStockSharesOutstanding |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionDescription of voting rights of nonredeemable preferred stock. Includes eligibility to vote and votes per share owned. Include also, if any, unusual voting rights. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(28))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 505
-SubTopic 10
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-3
| Name: |
us-gaap_PreferredStockVotingRights |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe cash inflow from the additional capital contribution to the entity. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 14
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-14
| Name: |
us-gaap_ProceedsFromIssuanceOfCommonStock |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe cash inflow associated with the amount received from entity's raising of capital via private rather than public placement. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 14
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-14
| Name: |
us-gaap_ProceedsFromIssuanceOfPrivatePlacement |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionCash received on stock transaction after deduction of issuance costs.
| Name: |
us-gaap_SaleOfStockConsiderationReceivedOnTransaction |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionThe number of shares issued or sold by the subsidiary or equity method investee per stock transaction.
| Name: |
us-gaap_SaleOfStockNumberOfSharesIssuedInTransaction |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of shares issued during the period as a result of the conversion of convertible securities. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 470
-SubTopic 20
-Name Accounting Standards Codification
-Section 50
-Paragraph 1E
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481139/470-20-50-1E
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 4: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 505
-SubTopic 10
-Section 50
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-3
Reference 5: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.29-30)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_StockIssuedDuringPeriodSharesConversionOfConvertibleSecurities |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of new stock issued during the period. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 505
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481004/946-505-50-2
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(4)(b))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-03(i)(1))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147479886/946-10-S99-3
Reference 5: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(28))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 6: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 7: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_StockIssuedDuringPeriodSharesNewIssues |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTotal number of shares issued during the period, including shares forfeited, as a result of Restricted Stock Awards. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
| Name: |
us-gaap_StockIssuedDuringPeriodSharesRestrictedStockAwardGross |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe gross value of stock issued during the period upon the conversion of convertible securities. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 210
-SubTopic 10
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02.29-31)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_StockIssuedDuringPeriodValueConversionOfConvertibleSecurities |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionEquity impact of the value of new stock issued during the period. Includes shares issued in an initial public offering or a secondary public offering. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://www.xbrl.org/2003/role/exampleRef
-Topic 946
-SubTopic 830
-Name Accounting Standards Codification
-Section 55
-Paragraph 11
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480167/946-830-55-11
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 205
-Name Accounting Standards Codification
-Section 45
-Paragraph 4
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480767/946-205-45-4
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 505
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481004/946-505-50-2
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 946
-SubTopic 220
-Name Accounting Standards Codification
-Section S99
-Paragraph 3
-Subparagraph (SX 210.6-09(4)(b))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483575/946-220-S99-3
Reference 6: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(28))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
Reference 7: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
Reference 8: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 210
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.5-02(29))
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480566/210-10-S99-1
| Name: |
us-gaap_StockIssuedDuringPeriodValueNewIssues |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
duration |
|
| X |
- DefinitionDescription of the reverse stock split arrangement. Also provide the retroactive effect given by the reverse split that occurs after the balance sheet date but before the release of financial statements. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 4
-Subparagraph (SAB Topic 4.C)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-4
| Name: |
us-gaap_StockholdersEquityReverseStockSplit |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_StatementEquityComponentsAxis=us-gaap_CommonStockMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementEquityComponentsAxis=SONN_PrefundedWarrantMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementEquityComponentsAxis=SONN_CommonStockWarrantsMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
dei_LegalEntityAxis=SONN_ChardanCapitalMarketsLLCMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_TypeOfArrangementAxis=SONN_SalesAgreementMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_ClassOfWarrantOrRightAxis=SONN_UnderwriterWarrantsMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
srt_StatementScenarioAxis=srt_ScenarioForecastMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
srt_TitleOfIndividualAxis=SONN_InvestorsMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
srt_TitleOfIndividualAxis=SONN_ChardanCapitalMarketsLLCMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementEquityComponentsAxis=SONN_CommonWarrantMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
dei_LegalEntityAxis=SONN_BTIGLLCMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_TypeOfArrangementAxis=SONN_SecuritiesPurchaseAgreementMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
srt_TitleOfIndividualAxis=SONN_AccreditedInvestorsMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementClassOfStockAxis=SONN_SeriesThreeConvertiblePreferredStockMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementClassOfStockAxis=SONN_SeriesFourConvertiblePreferredStockMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementClassOfStockAxis=SONN_SeriesThreeWarrantsMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_SubsidiarySaleOfStockAxis=us-gaap_PrivatePlacementMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_TypeOfArrangementAxis=SONN_TwentyTwentyTwoSalesAgeementMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
dei_LegalEntityAxis=SONN_BTIGMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.23.3
| X |
- DefinitionAmount of noncash expense for share-based payment arrangement. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 45
-Paragraph 28
-Subparagraph (a)
-SubTopic 10
-Topic 230
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482740/230-10-45-28
| Name: |
us-gaap_ShareBasedCompensation |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 35
-Paragraph 1D
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480483/718-10-35-1D
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 35
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480483/718-10-35-3
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(3)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(02)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(03)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(02)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(03)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 20: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (d)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 21: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (d)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 22: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (e)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 23: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (e)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 24: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 25: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 26: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 27: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(iv)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 28: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(v)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_AwardTypeAxis=us-gaap_RestrictedStockUnitsRSUMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_IncomeStatementLocationAxis=us-gaap_ResearchAndDevelopmentExpenseMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_IncomeStatementLocationAxis=us-gaap_GeneralAndAdministrativeExpenseMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.23.3
Schedule of Restricted Stock Units Activity (Details) - Restricted Stock Units (RSUs) [Member] - $ / shares
|
12 Months Ended |
Sep. 30, 2023 |
Sep. 30, 2022 |
| Share-Based Compensation Arrangement by Share-Based Payment Award [Line Items] |
|
|
| Unvested beginning balance |
2,162
|
1,179
|
| Unvested weighted average grant fair value, beginning balance |
$ 175.56
|
$ 1,068.32
|
| Restricted stock unit, granted |
7,840
|
2,103
|
| Weighted average grant fair value, granted |
$ 21.47
|
$ 155.54
|
| Restricted stock unit, Vested |
(2,162)
|
(1,096)
|
| Weighted average grant fair value, Vested |
$ 12.55
|
$ 1,091.20
|
| Restricted stock unit, Forfeited |
(5,514)
|
(24)
|
| Weighted average grant fair value, Forfeited |
$ 21.34
|
$ 1,118.04
|
| Unvested ending balance |
2,326
|
2,162
|
| Unvested weighted average grant fair value, ending balance |
$ 21.78
|
$ 175.56
|
| X |
- DefinitionThe number of equity-based payment instruments, excluding stock (or unit) options, that were forfeited during the reporting period. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(03)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardEquityInstrumentsOtherThanOptionsForfeitedInPeriod |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionWeighted average fair value as of the grant date of equity-based award plans other than stock (unit) option plans that were not exercised or put into effect as a result of the occurrence of a terminating event. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(03)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardEquityInstrumentsOtherThanOptionsForfeituresWeightedAverageGrantDateFairValue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe number of grants made during the period on other than stock (or unit) option plans (for example, phantom stock or unit plan, stock or unit appreciation rights plan, performance target plan). Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardEquityInstrumentsOtherThanOptionsGrantsInPeriod |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe weighted average fair value at grant date for nonvested equity-based awards issued during the period on other than stock (or unit) option plans (for example, phantom stock or unit plan, stock or unit appreciation rights plan, performance target plan). Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardEquityInstrumentsOtherThanOptionsGrantsInPeriodWeightedAverageGrantDateFairValue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe number of non-vested equity-based payment instruments, excluding stock (or unit) options, that validly exist and are outstanding as of the balance sheet date. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardEquityInstrumentsOtherThanOptionsNonvestedNumber |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionPer share or unit weighted-average fair value of nonvested award under share-based payment arrangement. Excludes share and unit options. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardEquityInstrumentsOtherThanOptionsNonvestedWeightedAverageGrantDateFairValue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionThe number of equity-based payment instruments, excluding stock (or unit) options, that vested during the reporting period. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(02)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardEquityInstrumentsOtherThanOptionsVestedInPeriod |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe weighted average fair value as of grant date pertaining to an equity-based award plan other than a stock (or unit) option plan for which the grantee gained the right during the reporting period, by satisfying service and performance requirements, to receive or retain shares or units, other instruments, or cash in accordance with the terms of the arrangement. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(02)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardEquityInstrumentsOtherThanOptionsVestedInPeriodWeightedAverageGrantDateFairValue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 35
-Paragraph 1D
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480483/718-10-35-1D
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 35
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480483/718-10-35-3
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(3)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(02)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(03)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(02)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(03)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 20: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (d)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 21: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (d)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 22: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (e)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 23: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (e)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 24: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 25: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 26: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 27: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(iv)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 28: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(v)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_AwardTypeAxis=us-gaap_RestrictedStockUnitsRSUMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.23.3
| X |
- DefinitionThe number of grants made during the period on other than stock (or unit) option plans (for example, phantom stock or unit plan, stock or unit appreciation rights plan, performance target plan). Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardEquityInstrumentsOtherThanOptionsGrantsInPeriod |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe weighted average fair value at grant date for nonvested equity-based awards issued during the period on other than stock (or unit) option plans (for example, phantom stock or unit plan, stock or unit appreciation rights plan, performance target plan). Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardEquityInstrumentsOtherThanOptionsGrantsInPeriodWeightedAverageGrantDateFairValue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe number of non-vested equity-based payment instruments, excluding stock (or unit) options, that validly exist and are outstanding as of the balance sheet date. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardEquityInstrumentsOtherThanOptionsNonvestedNumber |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionPer share or unit weighted-average fair value of nonvested award under share-based payment arrangement. Excludes share and unit options. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardEquityInstrumentsOtherThanOptionsNonvestedWeightedAverageGrantDateFairValue |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
dtr-types:perShareItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 35
-Paragraph 1D
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480483/718-10-35-1D
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 35
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480483/718-10-35-3
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(3)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(02)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(03)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(02)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(03)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 20: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (d)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 21: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (d)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 22: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (e)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 23: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (e)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 24: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 25: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 26: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 27: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(iv)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 28: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(v)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_AwardTypeAxis=SONN_RestrictedStockAwardsMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.23.3
Share-Based Compensation (Details Narrative) - USD ($)
|
1 Months Ended |
12 Months Ended |
|
Jan. 31, 2023 |
Dec. 31, 2022 |
Dec. 31, 2021 |
Mar. 31, 2021 |
Jul. 31, 2020 |
Sep. 30, 2023 |
Sep. 30, 2022 |
Jan. 01, 2023 |
| Restricted Stock Units (RSUs) [Member] |
|
|
|
|
|
|
|
|
| Share-Based Compensation Arrangement by Share-Based Payment Award [Line Items] |
|
|
|
|
|
|
|
|
| Restricted stock units and vesting rights, description |
|
In December of 2022,
7,840 RSUs were granted, 100% of which vest on January 1, 2024
|
In December of 2021, 2,103 RSUs were granted, 100% of which vested on January 1, 2023
|
In March of 2021, an additional 152 RSUs were granted, 50% of which vested on March 25, 2022 and the remaining
50% vested on March 25, 2023
|
In
July of 2020, 2,115 restricted stock units (“RSUs”) were granted, 50% of which vested on April 2, 2021 and the remaining
50% vested on April 2, 2022
|
|
|
|
| Number of shares granted |
|
7,840
|
2,103
|
152
|
2,115
|
|
|
|
| Restricted stock, shares issued |
5,514
|
|
|
|
|
|
|
|
| Stock compensation modification, incremental expense |
$ 38,837
|
|
|
|
|
|
|
|
| Unrecognized compensation expense |
|
|
|
|
|
$ 12,194
|
|
|
| Restricted Stock Units (RSUs) [Member] | Maximum [Member] |
|
|
|
|
|
|
|
|
| Share-Based Compensation Arrangement by Share-Based Payment Award [Line Items] |
|
|
|
|
|
|
|
|
| Weighted average period |
|
|
|
|
|
3 months
|
|
|
| Restricted Stock Awards (RSA) [Member] |
|
|
|
|
|
|
|
|
| Share-Based Compensation Arrangement by Share-Based Payment Award [Line Items] |
|
|
|
|
|
|
|
|
| Unrecognized compensation expense |
|
|
|
|
|
$ 37,812
|
|
|
| Restricted Stock Awards (RSA) [Member] | Maximum [Member] |
|
|
|
|
|
|
|
|
| Share-Based Compensation Arrangement by Share-Based Payment Award [Line Items] |
|
|
|
|
|
|
|
|
| Weighted average period |
|
|
|
|
|
3 months
|
|
|
| Common Stock [Member] |
|
|
|
|
|
|
|
|
| Share-Based Compensation Arrangement by Share-Based Payment Award [Line Items] |
|
|
|
|
|
|
|
|
| Restricted stock, shares issued |
|
|
|
|
|
|
1,087
|
|
| 2020 Omnibus Equity Incentive Plan [Member] | Common Stock [Member] |
|
|
|
|
|
|
|
|
| Share-Based Compensation Arrangement by Share-Based Payment Award [Line Items] |
|
|
|
|
|
|
|
|
| Number of shares available for issuance |
|
|
|
|
|
14,480
|
|
14,480
|
| X |
- DefinitionWeighted-average period over which cost not yet recognized is expected to be recognized for award under share-based payment arrangement, in 'PnYnMnDTnHnMnS' format, for example, 'P1Y5M13D' represents reported fact of one year, five months, and thirteen days. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_EmployeeServiceShareBasedCompensationNonvestedAwardsTotalCompensationCostNotYetRecognizedPeriodForRecognition1 |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:durationItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount of cost to be recognized for nonvested award under share-based payment arrangement. Excludes share and unit options. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_EmployeeServiceShareBasedCompensationNonvestedAwardsTotalCompensationCostNotYetRecognizedShareBasedAwardsOtherThanOptions |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionDescription of terms of share-based payment arrangement. Includes, but is not limited to, type of award or grantee and reason for issuance. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 718
-SubTopic 10
-Section 50
-Paragraph 1
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-1
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 718
-SubTopic 10
-Section 50
-Paragraph 2
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardDescription |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 35
-Paragraph 1D
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480483/718-10-35-1D
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 35
-Paragraph 3
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480483/718-10-35-3
Reference 3: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 4: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 5: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(3)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 6: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 7: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 8: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 9: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 10: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 11: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(02)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 12: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(03)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 13: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 14: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 15: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 16: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 17: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(01)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 18: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(02)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 19: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(2)(iii)(03)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 20: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (d)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 21: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (d)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 22: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (e)(1)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 23: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (e)(2)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 24: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(i)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 25: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(ii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 26: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 27: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(iv)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
Reference 28: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (f)(2)(v)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNet number of non-option equity instruments granted to participants. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)(1)(iv)(1)
-SubTopic 10
-Topic 718
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardNonOptionEquityInstrumentsGranted |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionNumber of shares authorized for issuance under share-based payment arrangement. Reference 1: http://www.xbrl.org/2003/role/exampleRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (a)(3)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardNumberOfSharesAuthorized |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionAn excess of the fair value of the modified award over the fair value of the award immediately before the modification. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 718
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (h)(2)(iii)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480429/718-10-50-2
| Name: |
us-gaap_ShareBasedCompensationArrangementByShareBasedPaymentAwardPlanModificationIncrementalCompensationCost |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionTotal number of shares issued during the period, including shares forfeited, as a result of Restricted Stock Awards. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-SubTopic 10
-Topic 505
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481112/505-10-50-2
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Topic 505
-SubTopic 10
-Name Accounting Standards Codification
-Section S99
-Paragraph 1
-Subparagraph (SX 210.3-04)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147480008/505-10-S99-1
| Name: |
us-gaap_StockIssuedDuringPeriodSharesRestrictedStockAwardGross |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:sharesItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_AwardTypeAxis=us-gaap_RestrictedStockUnitsRSUMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
srt_RangeAxis=srt_MaximumMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_AwardTypeAxis=SONN_RestrictedStockAwardsMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_StatementEquityComponentsAxis=us-gaap_CommonStockMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
| X |
- Details
| Name: |
us-gaap_PlanNameAxis=SONN_TwentyTwentyOmnibusEquityIncentivePlanMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.23.3
Schedule of Deferred Tax Assets and Liabilities (Details) - USD ($)
|
Sep. 30, 2023 |
Sep. 30, 2022 |
| Income Tax Disclosure [Abstract] |
|
|
| Net operating loss carryforwards |
$ 27,996,751
|
$ 25,858,311
|
| Research and development credit carryforwards |
3,106,675
|
2,151,942
|
| Amortization |
4,692,227
|
2,897,388
|
| Share-based compensation |
226
|
66,832
|
| Operating lease liability |
57,319
|
71,748
|
| Accrued expenses and other |
546,612
|
314,210
|
| Section 163(j) disallowed interest expense |
763,172
|
|
| Property and equipment |
|
4,790
|
| Gross deferred tax assets |
37,162,982
|
31,365,221
|
| Less: valuation allowance |
(37,100,582)
|
(31,293,092)
|
| Deferred tax assets |
62,400
|
72,129
|
| Property and equipment |
(7,954)
|
|
| Operating lease right-of-use asset |
(54,446)
|
(72,129)
|
| Net deferred tax assets |
|
|
| X |
- DefinitionDeferred tax assets amortization
| Name: |
SONN_DeferredTaxAssetsAmortization |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionDeferred tax assets operting lease liability
| Name: |
SONN_DeferredTaxAssetsOpertingLeaseLiability |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionDeferred tax assets tax deferred expense section disallowed interest expense.
| Name: |
SONN_DeferredTaxAssetsTaxDeferredExpenseSectionDisallowedInterestExpense |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionDeferred tax liabilities right of use asset
| Name: |
SONN_DeferredTaxLiabilitiesRightOfUseAsset |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount before allocation of valuation allowances of deferred tax asset attributable to deductible temporary differences and carryforwards. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (b)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-2
| Name: |
us-gaap_DeferredTaxAssetsGross |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount, after allocation of valuation allowances and deferred tax liability, of deferred tax asset attributable to deductible differences and carryforwards, without jurisdictional netting. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-2
| Name: |
us-gaap_DeferredTaxAssetsLiabilitiesNet |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount after allocation of valuation allowances of deferred tax asset attributable to deductible temporary differences and carryforwards. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-2
| Name: |
us-gaap_DeferredTaxAssetsNet |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount before allocation of valuation allowances of deferred tax asset attributable to deductible operating loss carryforwards. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-6
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 740
-SubTopic 10
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-8
| Name: |
us-gaap_DeferredTaxAssetsOperatingLossCarryforwards |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount before allocation of valuation allowances of deferred tax asset attributable to deductible temporary differences from property, plant, and equipment.
| Name: |
us-gaap_DeferredTaxAssetsPropertyPlantAndEquipment |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount before allocation of valuation allowances of deferred tax asset attributable to deductible research tax credit carryforwards. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-6
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 740
-SubTopic 10
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-8
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 740
-SubTopic 10
-Section 50
-Paragraph 3
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-3
| Name: |
us-gaap_DeferredTaxAssetsTaxCreditCarryforwardsResearch |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount before allocation of valuation allowances of deferred tax asset attributable to deductible temporary differences from share-based compensation. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-6
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 740
-SubTopic 10
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-8
| Name: |
us-gaap_DeferredTaxAssetsTaxDeferredExpenseCompensationAndBenefitsShareBasedCompensationCost |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount, before allocation of valuation allowance, of deferred tax asset attributable to deductible temporary differences from reserves and accruals, classified as other. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-6
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 740
-SubTopic 10
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-8
| Name: |
us-gaap_DeferredTaxAssetsTaxDeferredExpenseReservesAndAccrualsOther |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of deferred tax assets for which it is more likely than not that a tax benefit will not be realized. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Subparagraph (c)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-2
| Name: |
us-gaap_DeferredTaxAssetsValuationAllowance |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of deferred tax liability attributable to taxable temporary differences from property, plant, and equipment. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-6
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 740
-SubTopic 10
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-8
| Name: |
us-gaap_DeferredTaxLiabilitiesPropertyPlantAndEquipment |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
credit |
| Period Type: |
instant |
|
v3.23.3
v3.23.3
Income Taxes (Details Narrative) - USD ($)
|
12 Months Ended |
|
Sep. 30, 2023 |
Sep. 30, 2022 |
Aug. 31, 2022 |
| Operating Loss Carryforwards [Line Items] |
|
|
|
| Net operating losses |
$ 4,800,000
|
|
|
| Tax credit carryforwards |
3,106,675
|
$ 2,151,942
|
|
| Increase in valuation allowance |
5,800,000
|
$ 8,300,000
|
|
| Excise tax |
|
|
1.00%
|
| Minimum [Member] |
|
|
|
| Operating Loss Carryforwards [Line Items] |
|
|
|
| Corporate alternative tax |
|
|
15.00%
|
| Domestic Tax Authority [Member] |
|
|
|
| Operating Loss Carryforwards [Line Items] |
|
|
|
| Net operating losses |
103,600,000
|
|
|
| Tax credit carryforwards |
2,400,000
|
|
|
| State and Local Jurisdiction [Member] |
|
|
|
| Operating Loss Carryforwards [Line Items] |
|
|
|
| Net operating losses |
69,500,000
|
|
|
| Tax credit carryforwards |
800,000
|
|
|
| Foreign Tax Authority [Member] |
|
|
|
| Operating Loss Carryforwards [Line Items] |
|
|
|
| Net operating losses |
$ 8,900,000
|
|
|
| Expiration period |
expire
in 2027
|
|
|
| X |
- DefinitionCorporate alternative tax percentage
| Name: |
SONN_CorporateAlternativeTaxPercentage |
| Namespace Prefix: |
SONN_ |
| Data Type: |
dtr-types:percentItemType |
| Balance Type: |
na |
| Period Type: |
instant |
|
| X |
- DefinitionOperating loss carry forwards expiration period.
| Name: |
SONN_OperatingLossCarryforwardsExpirationPeriod |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAmount before allocation of valuation allowances of deferred tax asset attributable to deductible research tax credit carryforwards. Reference 1: http://www.xbrl.org/2009/role/commonPracticeRef
-Topic 740
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 6
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-6
Reference 2: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 740
-SubTopic 10
-Section 50
-Paragraph 8
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-8
Reference 3: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 740
-SubTopic 10
-Section 50
-Paragraph 3
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-3
| Name: |
us-gaap_DeferredTaxAssetsTaxCreditCarryforwardsResearch |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionAmount of operating loss carryforward, before tax effects, available to reduce future taxable income under enacted tax laws. Reference 1: http://fasb.org/us-gaap/role/ref/legacyRef
-Name Accounting Standards Codification
-Topic 740
-SubTopic 10
-Section 50
-Paragraph 3
-Subparagraph (a)
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147482685/740-10-50-3
| Name: |
us-gaap_OperatingLossCarryforwards |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
instant |
|
| X |
- DefinitionLine items represent financial concepts included in a table. These concepts are used to disclose reportable information associated with domain members defined in one or many axes to the table.
| Name: |
us-gaap_OperatingLossCarryforwardsLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
srt_RangeAxis=srt_MinimumMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
v3.23.3
| X |
- DefinitionProceeds from sale of state net operating losses.
| Name: |
SONN_ProceedsFromSaleOfStateNetOperatingLosses |
| Namespace Prefix: |
SONN_ |
| Data Type: |
xbrli:monetaryItemType |
| Balance Type: |
debit |
| Period Type: |
duration |
|
| X |
- DefinitionDetail information of subsequent event by type. User is expected to use existing line items from elsewhere in the taxonomy as the primary line items for this disclosure, which is further associated with dimension and member elements pertaining to a subsequent event. Reference 1: http://www.xbrl.org/2003/role/disclosureRef
-Topic 830
-SubTopic 30
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147481674/830-30-50-2
Reference 2: http://www.xbrl.org/2003/role/disclosureRef
-Topic 855
-SubTopic 10
-Name Accounting Standards Codification
-Section 50
-Paragraph 2
-Publisher FASB
-URI https://asc.fasb.org//1943274/2147483399/855-10-50-2
| Name: |
us-gaap_SubsequentEventLineItems |
| Namespace Prefix: |
us-gaap_ |
| Data Type: |
xbrli:stringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Details
| Name: |
us-gaap_SubsequentEventTypeAxis=us-gaap_SubsequentEventMember |
| Namespace Prefix: |
|
| Data Type: |
na |
| Balance Type: |
|
| Period Type: |
|
|
Grafico Azioni Sonnet BioTherapeutics (NASDAQ:SONN)
Storico
Da Feb 2025 a Mar 2025

Grafico Azioni Sonnet BioTherapeutics (NASDAQ:SONN)
Storico
Da Mar 2024 a Mar 2025

