As
filed with the U.S. Securities and Exchange Commission on January 19, 2024
Registration
No. 333-
UNITED
STATES
SECURITIES
AND EXCHANGE COMMISSION
Washington,
D.C. 20549
FORM
S-1
REGISTRATION
STATEMENT
UNDER
THE
SECURITIES ACT OF 1933
AZITRA,
INC.
(Exact
name of registrant as specified in its charter)
| Delaware |
|
2834 |
|
46-4478536 |
(State
or other jurisdiction of
incorporation
or organization) |
|
(Primary
Standard Industrial
Classification
Code Number) |
|
(I.R.S.
Employer
Identification
Number) |
21
Business Park Drive
Branford,
CT 06405
(203)
646-6446 |
(Address,
including zip code, and telephone number, including area code, of registrant’s principal executive offices)
Francisco
D. Salva
21
Business Park Drive
Branford,
CT 06405
(203)
646-6446
(Name,
address, including zip code, and telephone number, including area code, of agent for service)
Copies
to:
Daniel
K. Donahue, Esq.
Greenberg
Traurig, LLP
18565
Jamboree Road, Suite 500
Irvine,
California 92612
(949)
732-6557 |
William
N. Haddad, Esq.
Arif
Soto, Esq.
Venable
LLP
151
W. 42nd Street, 49th Floor
New
York, New York 10036
(212)
307-5598 |
Approximate
date of commencement of proposed sale to the public: As soon as practicable after this Registration Statement becomes effective.
If
any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the
Securities Act of 1933, check the following box. ☒
If
this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the
following box and list the Securities Act registration statement number of the earlier effective registration statement for the same
offering. ☐
If
this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the
Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If
this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the
Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
Indicate
by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company,
or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller
reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act:
| Large
accelerated filer ☐ |
Accelerated
filer ☐ |
Non-accelerated
filer ☒ |
Smaller
reporting company ☒ |
| |
|
|
Emerging
Growth Company ☒ |
| |
|
|
|
If
an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying
with any new or revised financial accounting standards provided to Section 7(a)(2)(B) of the Securities Act. ☐
The
Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the
Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective
in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the Registration Statement shall become effective
on such date as the Commission, acting pursuant to said Section 8(a), may determine.
The
information contained in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration
statement filed with the U.S. Securities and Exchange Commission is declared effective. This preliminary prospectus is not an offer to
sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
| PRELIMINARY PROSPECTUS |
SUBJECT TO COMPLETION |
DATED JANUARY 19,
2024 |
Up
to 3,000,000 Shares of Common
Stock
Up
to 3,000,000 Pre-Funded Warrants to purchase up to
3,000,000 Shares of Common Stock

Azitra,
Inc.
This
is a firm commitment public offering of 3,000,000 shares of common stock, par value $0.0001 per share, of Azitra, Inc. The final
public offering price will be determined through negotiation between us and the lead underwriter in the offering and the recent market
prices used throughout this prospectus may not be indicative of the final offering price.
We
are also offering pre-funded warrants, or Pre-Funded Warrants, to purchase 3,000,000 shares of our common stock to those purchasers
whose purchase of common stock in this offering would otherwise result in the purchaser, together with its affiliates and certain
related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding shares of our
common stock immediately following the consummation of this offering, in lieu of shares of common stock that would otherwise result
in such purchaser’s beneficial ownership exceeding 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding
shares of common stock. The purchase price of each Pre-Funded Warrant will be equal to the public offering price per share of common
stock sold in this offering minus $0.001, the exercise price per share of common stock of each Pre-Funded Warrant. The Pre-Funded
Warrants are immediately exercisable and may be exercised at any time until all of the Pre-Funded Warrants are exercised in full.
For each Pre-Funded Warrant we sell, the number of shares of common stock we are offering will be decreased on a one-for-one
basis.
Our
common stock is listed on the NYSE American, under the symbol “AZTR.” On January 18, 2024, the closing sale of our common
stock was $2.05. There is currently no established trading market for the offered Pre-Funded Warrants and we do not expect one to develop.
Without an active trading market, the liquidity of the Pre-Funded Warrants will be limited.
We
are an “emerging growth company” under the U.S. federal securities laws and have elected to comply with certain reduced public
company reporting requirements.
Investing
in our shares of common stock involves a high degree of risk. See the section titled “Risk Factors” beginning on page
11. Neither the Securities and Exchange Commission, or the SEC, nor any state securities commission has approved or disapproved of these
securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
| | |
Per Share | | |
Per Pre-Funded Warrant | | |
Total | |
| Public offering price | |
$ | | | |
$ | | | |
$ | | |
| Underwriting discounts and commissions(1) | |
$ | | | |
$ | | | |
$ | | |
| Proceeds to us, before expenses | |
$ | | | |
$ | | | |
$ | | |
| (1) | Underwriting
discounts and commissions do not include a non-accountable expense allowance equal to 1%
of the public offering price payable to the underwriters. We refer you to “Underwriting”
beginning on page 107 for additional information regarding underwriters’ compensation. |
We
have granted a 45-day option to the representative of the underwriters to purchase up to 450,000 additional shares of
common stock (and/or Pre-Funded Warrants to purchase up to 450,000 shares of common stock in lieu thereof) solely to cover
over-allotments, if any.
The
underwriters expect to deliver the shares to purchasers on or about , 2024.
ThinkEquity
The
date of this prospectus is , 2024.

Table
of Contents
You
should rely only on the information contained in this prospectus. We have not authorized any other person to provide you with information
different from or in addition to that contained in this prospectus, and we take no responsibility for any other information others may
give you. If anyone provides you with different or inconsistent information, you should not rely on it. We are not making an offer to
sell these securities in any jurisdiction where an offer or sale is not permitted. You should assume that the information appearing in
this prospectus is accurate only as of the date on the front cover of this prospectus. Our business, financial condition, results of
operations and prospects may have changed since that date.
No
action is being taken in any jurisdiction outside the United States to permit a public offering of our common stock or possession or
distribution of this prospectus in that jurisdiction. Persons who come into possession of this prospectus in jurisdictions outside the
United States are required to inform themselves about and to observe any restrictions as to this offering and the distribution of this
prospectus applicable to that jurisdiction.
As
used in this prospectus, unless the context indicates or otherwise requires, “the Company,” “our Company,” “we,”
“us,” and “our” refer to Azitra, Inc., a Delaware corporation.
INDUSTRY
AND MARKET DATA
This
prospectus, particularly the section “Business,” contains observations, statistical data, estimates, and forecasts that are
based on independent industry, government and non-government organization publications or other publicly available information, as well
as other information based on our internal sources. Although we believe that the third-party sources referred to in this prospectus are
reliable, estimates as they relate to projections involve numerous assumptions, are subject to risks and uncertainties, and are subject
to change based on various factors, including those discussed under the section titled “Risk Factors” and elsewhere
in this prospectus. These and other factors could cause results to differ materially from those expressed in the estimates made by the
independent parties and by us.
Certain
information in the text of this prospectus is contained in independent industry government and non-governmental organizational publications.
The sources of these publications are provided below:
| |
● |
Stacy
and Belkaid Study, Apollo Stacy and Yasmine Belkaid, Microbial Guardians of Skin Health. Science, 2019 Jan 18;363(6424):227-228.
Doi: 10.1126/science.aat4326. PMID: 30655428 |
| |
● |
Oh
Study, Zhou W, Spoto M, Hardy R, Guan C, Fleming E, Larson PJ, Brown JS, Oh J. Host-Specific Evolutionary and Transmission Dynamics
Shape the Functional Diversification of Staphylococcus epidermidis in Human Skin. Cell. 2020 Feb 6;180(3):454-470.e18. doi: 10.1016/j.cell.2020.01.006.
Epub 2020 Jan 30. PMID: 32004459; PMCID |
| |
● |
Satoh
Study, Satoh TK, Mellett M, Meier-Schiesser B, Fenini G, Otsuka A, Beer HD, Rordorf T, Maul JT, Hafner J, Navarini AA, Contassot
E, French LE. IL-36γ drives skin toxicity induced by EGFR/MEK inhibition and commensal Cutibacterium acnes. J Clin Invest.
2020 Mar 2;130(3):1417-1430. doi: 10.1172/JCI128678. PMID: 31805013; PMCID: PMC7269569 |
| |
● |
Barbati
Study, Netherton Syndrome in Children: Management and Future Perspectives, Federica Barbati, Mattia Giovannini Teresa Oranges, Lorenzo
Lodi, Simona Barni, Elio Novembre, Ermanno Baldo, Mario Cristofolini, Stefano Stagi, Silvia Ricci, Francesca Mori, Cesare Filippeschi,
Chiara Azzari and Giuseppe Indol; Frontiers in Pediatrics, May 2021 |
| |
● |
Sun
Study, Netherton syndrome: A case report and review of the literature, Joannie D. Sun, MD, and Kenneth G. Linden, PhD, MD, International
Journal of Dermatology 2006 |
| |
● |
Orphanet,
Netherton Syndrome, Orphanet: Netherton syndrome |
PROSPECTUS
SUMMARY
This
summary highlights certain information appearing elsewhere in this prospectus. Investing in our common stock involves a high degree of
risk. Because it is only a summary, it does not contain all of the information that you should consider before investing in our common
stock and it is qualified in its entirety by, and should be read in conjunction with, the more detailed information appearing elsewhere
in this prospectus. Before you decide to invest in our common stock, you should read the entire prospectus carefully, including “Risk
Factors” beginning on page 11 and the financial statements and related notes included in this prospectus.
On
May 17, 2023, we effected a forward stock split at a ratio of 7.1-for-1. All share and share price information in this prospectus have
been adjusted to give effect to the forward stock split.
Our
Company
We
are an early-stage clinical biopharmaceutical company focused on developing innovative therapies for precision dermatology using engineered
proteins and topical live biotherapeutic products. We have built a proprietary platform that includes a microbial library comprised of
approximately 1,500 unique bacterial strains that can be screened for unique therapeutic characteristics. The platform is augmented by
an artificial intelligence and machine learning technology that analyzes, predicts and helps screen our library of strains for drug like
molecules. The platform also utilizes a licensed genetic engineering technology, which can enable the transformation of previously genetically
intractable strains. Our initial focus is on the development of genetically engineered strains of Staphylococcus epidermidis, or S.
epidermidis, which we consider to be an optimal therapeutic candidate species for engineering of dermatologic therapies. The particular
species demonstrates a number of well-described properties in the skin. As of the date of this prospectus, we have identified among our
microbial library over 60 distinct bacterial species that we believe are capable of being engineered to create living organisms or engineered
proteins with significant therapeutic effect.
We
are a pioneer in genetically engineering bacteria for therapeutic use in dermatology. Our goal is to leverage our platforms and internal
microbial library bacterial strains to create new therapeutics that are either engineered living organisms or engineered proteins or
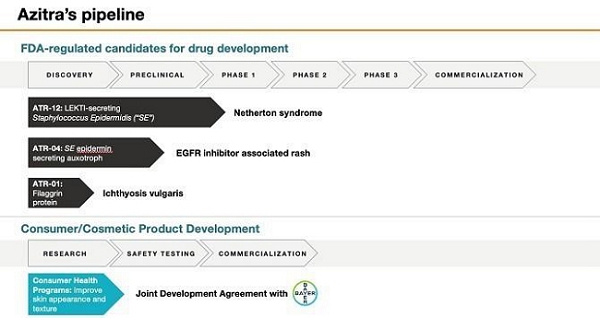
peptides to treat skin diseases. Our initial focus is on the development of our current product candidates, including:
| |
● |
ATR-12,
a genetically modified strain of S. epidermidis for treating the orphan disease, Netherton syndrome, a chronic and sometimes
fatal disease of the skin estimated to affect approximately one to nine in every 100,000, but its prevalence may be underestimated
due to misdiagnosis caused by similarities to other skin diseases. We received Pediatric Rare Disease Designation for ATR-12 by the
United States Food and Drug Administration, or FDA, in 2019. In December 2022, we submitted an investigational new drug application,
or IND, for a Phase 1b clinical trial of ATR-12 in Netherton syndrome patients, and on January 27, 2023, we received notification
from the FDA that the “study may proceed” with respect to the proposed Phase 1b clinical trial. We commenced our Phase
1b clinical trial in December 2023 and expect to report initial safety results in the second half of 2024. |
| |
● |
ATR-04,
a genetically modified strain of S. epidermidis for treating the papulopustular rash experienced by cancer patients undergoing
epidermal growth factor receptor inhibitor, or EGFRi, targeted therapy. We intend to submit an IND for a Phase 1b clinical trial
in certain cancer patients undergoing EGFRi targeted therapy by the first half of 2024. Subject to FDA clearance of our IND, we expect
to commence our Phase 1b clinical trial in the second half of 2024. |
| |
● |
ATR-01,
an engineered recombinant human filaggrin protein for treating ichthyosis vulgaris, a chronic, xerotic (abnormally dry), scaly skin
disease with an estimated incidence and prevalence of 1 in 250, which suggests a total patient population of 1.3 million in the United
States. We are planning to complete lead optimization and IND-enabling studies in 2024 to support an IND filing target in
mid-2025. |
| |
● |
Two
separate strains of bacterial microbes are being investigated and developed by us and Bayer Consumer Care AG, the consumer products
division of Bayer AG, or Bayer, the international life science company pursuant to a Joint Development Agreement, or JDA, entered
into between the parties in December 2019. Under the terms of the JDA, we are responsible for testing our library of bacterial strains
and their natural products for key preclinical properties. After screening through hundreds of strains, we and Bayer have selected
two particular strains to move forward into further development. Bayer holds the exclusive option to license the patent rights to
these strains. In December 2020, Bayer purchased $8 million of our Series B preferred stock, which converted into 1,449,743 shares
of our common stock, representing approximately 12.0% of our outstanding shares of common stock. |
We
also have established partnerships with teams from Carnegie Mellon University and the Fred Hutchinson Cancer Center, or Fred Hutch, two
of the premier academic centers in the United States. Our collaboration with the Carnegie Mellon based team takes advantage of the power
of whole genome sequencing. This partnership is mining our proprietary library of bacterial strains for novel, drug-like peptides and
proteins. The artificial intelligence/machine learning technology developed by this team predicts the molecules made by microbes from
their genetic sequences. The system then compares the predictions to the products actually made through tandem mass spectroscopy and/or
nuclear magnetic resonance imaging to refine future predictions. The predictions can be compared to publicly available 2D and 3D protein
databases to select drug like structures.
We
hold an exclusive, worldwide license from Fred Hutch regarding the use of its patented SyngenicDNA Minicircle Plasmid, or SyMPL, technologies
for all fields of genetic engineering, including to discover, develop and commercialize engineered microbial therapies and microbial-derived
peptides and proteins for skin diseases. We are utilizing our licensed patent rights to build plasmids in order to make genetic transformations
that have never been previously achieved. Our collaboration with Fred Hutch is led by Dr. Christopher Johnston, an expert in microbial
engineering, and the innovator behind the SyMPL technology.
Bayer
Partnership
In
December 2019, we entered into a Joint Development Agreement, or JDA, with Bayer pursuant to which we agreed to the joint development
of certain strains selected from our proprietary microbial library. We and Bayer have agreed to cooperate in the identification and in
vitro and ex vivo characterization of microbial strains for topical formulations, which we intend to develop as potential
over-the-counter cosmetic products. Bayer paid us a one-time payment upon execution of the JDA and has agreed to reimburse us for our
development costs. In October 2021, Bayer expanded the option agreement and paid us a second fee for additional characterization work.
We have granted Bayer an option to acquire an exclusive royalty bearing license for up to six strains subject to development activities
under the JDA, including an exclusive royalty bearing license to any related patent rights. Bayer has an option to acquire the exclusive
license rights for a period of six months following our delivery of the results of the JDA development activities to Bayer. After screening
through hundreds of strains, we and Bayer have selected two particular strains to move forward with in vitro and ex vivo characterization.
In
September 2020, Bayer’s venture capital group, LEAPS by Bayer, purchased $8 million of our Series B preferred stock, which converted
into 1,449,743 shares of our common stock, representing approximately 12.0% of our outstanding shares of common stock.
Our
Strategy
Beyond
our three lead product candidates and collaboration with Bayer, our goal is to develop a broad portfolio of product candidates focused
on expanding the application of our platforms for precision dermatology. We believe that we have established a unique position in advancing
the development of biologics for precision dermatology.
We
intend to create a broad portfolio of product candidates for precision dermatology through our development of genetically engineered
proteins selected from our proprietary microbial library of approximately 1,500 unique bacterial strains. Our strategy is as follows:
| |
● |
Build
a sustainable precision dermatology company. Our goal is to build a leading precision dermatology company with a sustainable
pipeline of product candidates. To that end, we are focused on rapidly advancing our current pipeline of live biotherapeutic candidates
while actively developing additional product candidates. Each of our current product candidates are proprietary and subject to pending
patent applications. We expect that most of our genetically engineered product candidates we develop will be eligible for patent
protection. |
| |
● |
Advance
our lead product candidates, ATR-12 and ATR-04, through clinical trials. We expect to report initial safety results
of our Phase 1b clinical trial for our ATR-12 in Netherton syndrome patients in the second half of 2024 and are currently
planning to commence a Phase 1b trial of our ATR-04 in certain cancer patients undergoing EGFRi therapy in the second half of 2024.
We have a cleared IND for ATR-12 and expect to file an IND for ATR-04 in the first half of 2024. |
| |
● |
Broaden
our platform by selectively exploring strategic partnerships that maximize the potential of our precision dermatology programs.
We intend to maintain significant rights to all of our core technologies and product candidates. However, we will continue to evaluate
partnering opportunities in which a strategic partner could help us to accelerate development of our technologies and product candidates,
provide access to synergistic combinations, or provide expertise that could allow us to expand into the treatment of different types
of skin diseases. We may also broaden the reach of our platform by selectively in-licensing technologies or product candidates. In
addition, we will consider potentially out-licensing certain of our proprietary technologies for indications and industries that
we are not pursuing. We believe our genetic engineering techniques and technologies have applicability outside of the field of medicine,
including cosmetics and in the generation of clean fuels and bioremediation. |
| |
● |
Leverage
our academic partnerships. We currently have partnerships with investigators at the Fred Hutchinson Cancer Center, Yale University,
Jackson Laboratory for Genomic Medicine, and Carnegie Mellon University. We expect to leverage these partnerships and potentially
expand them or form other academic partnerships to bolster our engineering platforms and expand our research and development pipeline. |
| |
● |
Expand
on our other potential product candidates. Beyond our three lead product candidates, our goal is to develop a broad portfolio
of product candidates focused on expanding the application of our platforms for precision dermatology. We have a proprietary platform
for discovering and developing therapeutic products for precision dermatology. Our platform is built around a microbial library comprised
of approximately 1,500 unique bacterial strains to allow screening for unique therapeutic characteristics and utilizes a microbial
genetic technology that analyzes, predicts and engineers the proteins, peptides and molecules made by skin microbes. Our ability
to genetically engineer intractable microbial species is uniquely leveraged by our exclusive license to the SyMPL technology. |
Our
Intellectual Property
As
of the date of this prospectus, we own or exclusively license three issued U.S. patents, 12 pending U.S. patent applications, three pending PCT application and 59 other foreign patents and patent applications that are important
to the development of our business.
Our
Leadership Team
We
are led by Francisco D. Salva, our chief executive officer, and Travis Whitfill, our co-founder and chief operating officer, who have
more than 35 years of combined experience in the management of biotechnology companies and healthcare investing. Mr. Salva was previously
a co-founder of Acerta Pharma, which was sold to AstraZeneca for approximately $6.3 billion in a staged acquisition in 2016. He also
worked on the turnaround of Pharmacyclics, which subsequently sold to Abbvie for approximately $21 billion in 2015. Before that, Mr.
Salva spent almost a decade in life sciences venture capital. Mr. Whitfill served as associate research scientist and serves as assistant
professor adjunct at Yale University with appointments in the Departments of Pediatrics and Emergency Medicine. He spent nearly a decade
in venture capital as a partner in a biotech-focused venture capital fund, Bios Partners. He has led numerous grant-funded projects,
holds nearly a dozen patents and has co-authored over 60 publications. Our board of directors, or Board, is comprised of renowned group
of senior executives, scientists and investors in the biotechnology industry.
Our
Competitive Strengths
We
are a pioneer in genetically engineering bacteria for therapeutic use in dermatology clinical trials. We have built a proprietary platform
that includes a microbial library comprised of approximately 1,500 unique bacterial strains that are screened for therapeutic characteristics
as well as lead drug candidates. Furthermore, we have exclusively licensed a novel technology, which potentially enables the genetic
transformation of previously intractable bacterial microbes. The history of recombinant protein engineering in biotech has traditionally
been limited to less than 20 species. Our licensed technology opens up the potential to genetically engineer thousands of microbial species
to build proteins and peptides that have never been previously built. Our management team has significant experience in discovering,
developing, manufacturing and commercializing therapeutics. The members of our leadership team have specialized expertise developed at
companies including Pharmacyclics, Acerta Pharma, Castle Creek Biosciences, VYNE Therapeutics (fka Menlo Therapeutics), Revance Therapeutics,
Biogen, Novartis and Connetics Corp.
Our
Market Opportunity
We
believe there are significant market opportunities to capture in each of our addressable markets. The dermatology market itself has shown
considerable growth over the last decade and is predicted to continue to grow. According to Vision Research Reports, the dermatology
drug market surpassed $17 billion in 2021 and is expected to grow at a compound annual growth rate of 8.8% through 2030. Our first product
candidate to emerge from our platform focuses on the orphan indication of Netherton syndrome. Based on the Barbati and Sun Studies, we
believe that this product candidate represents a potential $250 million global sales opportunity by mid-2030. Our second product candidate
focuses on papulopustular rash due to EGFR inhibitors. We believe this product candidate represents a potential $1 billion global sales
opportunity by 2030. The diseases we intend to target are well characterized, often by a monogenic genetic mutation. Additionally, the
era of genomic sequencing has ushered in unprecedented progress in genetic testing. The defined molecular pathophysiology of over 100
rare skin diseases has now been defined.
Our
Corporate Information
We
were incorporated under the laws of the state of Delaware on January 2, 2014. Our principal executive offices are located at 21 Business
Park Drive, Branford, Connecticut 06405, and our telephone number is (203) 646-6446. Our website address is www.azitrainc.com. The information
contained in, or accessible through, our website is not incorporated by reference into this prospectus, and you should not consider any
information contained in, or that can be accessed through, our website as part of this prospectus or in deciding whether to purchase
our common stock or Pre-Funded Warrants.
We
own U.S. and foreign registered trademarks, including our company name. All other trademarks or trade names referred to in this prospectus
are the property of their respective owners. Solely for convenience, the trademarks and trade names in this prospectus are referred to
without the symbols ® and ™, but such references should not be construed as any indication that their respective owners will
not assert, to the fullest extent under applicable law, their rights thereto.
Private
Placements of our Convertible Securities
Prior
to our June 2023 initial public offering, or IPO, we have capitalized our operations through a series of private placements of our convertible
preferred stock and convertible promissory notes, for the total gross proceeds of $39 million, all of which were converted into a total
of 8,951,526 shares of our common stock upon the consummation of our IPO.
Initial
Public Offering
In
June 2023, the Company completed its IPO in which it issued and sold 1,500,000 shares of its common stock at a price to the public of
$5.00 per share. The shares began trading on the NYSE American on June 16, 2023 under the symbol “AZTR.” The net proceeds
received by the Company from the offering were $6.5 million, after deducting underwriting discounts, commissions and other offering expenses.
Changes
to our Capitalization
On
May 17, 2023, we amended and restated our certificate of incorporation to (i) increase our authorized common stock from 1,950,000 shares
to 100,000,000 shares, (ii) change the par value of our capital stock from $0.01 to $0.0001, (iii) change our corporate name from “Azitra
Inc” to “Azitra, Inc.” and (iv) effect a 7.1-for-1 forward stock split of our outstanding common stock, or the Stock
Split.
Except
as otherwise indicated herein, all share and share price information in this prospectus have been adjusted to give effect to the Stock
Split.
Implications
of Being an Emerging Growth Company
The
Jumpstart Our Business Startups Act, or the JOBS Act, was enacted in April 2012 with the intention of encouraging capital formation in
the United States and reducing the regulatory burden on newly public companies that qualify as “emerging growth companies.”
We are an emerging growth company within the meaning of the JOBS Act. As an emerging growth company, we may take advantage of certain
exemptions from various public reporting requirements, including:
| |
● |
the
requirement that our internal control over financial reporting be attested to by our independent registered public accounting firm
pursuant to Section 404 of the Sarbanes-Oxley Act of 2002; |
| |
● |
certain
requirements related to the disclosure of executive compensation in this prospectus and in our periodic reports and proxy statements; |
| |
● |
the
requirement that we hold a nonbinding advisory vote on executive compensation and any golden parachute payments; and |
| |
● |
the
ability to delay compliance with new or revised financial accounting standards until private companies are required to comply with
the new or revised financial accounting standard. |
We
may take advantage of the exemptions under the JOBS Act discussed above until we are no longer an emerging growth company. We will remain
an emerging growth company until the earliest to occur of (1) the last day of the fiscal year in which we have $1.235 billion or more
in annual revenue; (2) the date we qualify as a “large accelerated filer,” with at least $700 million of equity securities
held by non-affiliates; (3) the date on which we have issued, in any three-year period, more than $1.0 billion in non-convertible debt
securities; or (4) the last day of the fiscal year ending after the fifth anniversary of the IPO.
We
may choose to take advantage of some, but not all, of the available benefits under the JOBS Act. We have chosen to take advantage of
all of the other exemptions discussed above. Accordingly, the information contained herein and in our subsequent filing with the SEC
may be different than the information you receive from other public companies in which you hold stock.
For
certain risks related to our status as an emerging growth company, see the disclosure elsewhere in this prospectus under “Risk
Factors—Risks Related to this Offering and Owning Our Common Stock—We are an ‘emerging growth company’ under
the JOBS Act and we cannot be certain if the reduced disclosure requirements applicable to emerging growth companies will make our common
stock less attractive to investors.”
Implications
of Being a Smaller Reporting Company
Additionally,
we are a “smaller reporting company” as defined in Rule 10(f)(1) of Regulation S-K. Smaller reporting companies may take
advantage of certain reduced disclosure obligations, including, among other things, providing only two years of audited financial statements.
We will remain a smaller reporting company until the last day of the fiscal year in which (1) the market value of our common stock held
by non-affiliates equals or exceeds $250 million as of the end of that year’s second fiscal quarter, or (2) our annual revenues
equaled or exceeded $100 million during such completed fiscal year and the market value of our common stock held by non-affiliates equals
or exceeds $700 million as of the end of that year’s second fiscal quarter.
THE
OFFERING
| Issuer |
Azitra,
Inc. |
| |
|
| Common
stock offered |
3,000,000
shares |
| |
|
| Pre-Funded Warrants offered |
We
are also offering, in lieu of shares of our common stock to certain purchasers, Pre-Funded
Warrants to purchase 3,000,000 shares of our common stock that would otherwise result in
such purchaser’s beneficial ownership exceeding 4.99% (or, at the election of the purchaser,
9.99%) of our outstanding shares of common stock. Each Pre-Funded Warrant will be exercisable
for one share of our common stock. This offering also relates to the shares of common stock
issuable upon exercise of any Pre-Funded Warrant sold in this offering. For each Pre-Funded
Warrant that we sell, the number of shares of common stock that we are offering will be decreased
on a one-for-one basis.
The purchase price of each Pre-Funded Warrant will equal the price
per share of common stock being sold to the public in this offering, minus $0.001, and the exercise price of each Pre-Funded Warrant will
be $0.001 per share.
The Pre-Funded Warrants will be exercisable immediately and may be
exercised at any time until all of the Pre-Funded Warrants are exercised in full. To better understand the terms of the Pre-Funded Warrants,
you should carefully read the “Description of Securities–Pre-Funded Warrants” section of this prospectus. You should
also read the form of Pre-Funded Warrant, which is filed as an exhibit to the registration statement of which this prospectus forms a
part.
|
| |
|
| Common
stock to be outstanding after this offering |
15,097,643 shares
(or 15,547,643 shares if the underwriters’ option to purchase additional shares is exercised in full) of common stock,
in each case, assuming no sales of Pre-Funded Warrants offered by us, which, to the extent Pre-Funded Warrants are sold, will reduce
the number of shares of common stock that we are offering on a one-for-one basis. |
| |
|
| Over-allotment
option |
We
have granted the underwriters a 45 day option from the date of this prospectus, exercisable one or more times in whole or in part,
to purchase up to an additional 450,000 shares of common stock and/or up to an additional 450,000 Pre-funded Warrants (15% of
the total number of shares of common stock and/or Pre-funded Warrants to be offered by us in the offering), solely to cover over-allotments,
if any. |
| |
|
| Use
of proceeds |
We
estimate that we will receive net proceeds of approximately $5.1 million from our sale of common stock in this offering, or
approximately $6.0 million if the underwriters exercise their over-allotment option in full, assuming a public offering
price of $2.05 per share, the last reported sale price of our common stock on The NYSE American on January 18, 2024.
We intend to use the net proceeds from this offering, along with our existing cash and cash equivalents, for clinical trials and
product development, research and development, clinical manufacturing as well as for working capital and other general corporate
purposes. See the section titled “Use of Proceeds” in this prospectus for a more complete description of the intended
use of processed from this offering. |
| |
|
| Trading
market and symbol |
Our
common stock is listed on NYSE American under the symbol “AZTR.”
We do not intend to list the Pre-Funded Warrants
on NYSE American or any other national securities exchange or nationally recognized trading system. Without an active trading market,
the liquidity of the Pre-Funded Warrants will be limited. |
| |
|
| Risk
factors |
Investing
in our common stock involves a high degree of risk. See the section titled “Risk Factors” beginning on page 11
and the other information in this prospectus for a discussion of the factors you should consider carefully before you decide to invest
in our common stock. |
| |
|
| Lock-up |
We
have agreed, subject to certain exceptions, not to sell, offer, agree to sell, contract to sell, hypothecate, pledge, grant any option
to purchase, make any short sale of, or otherwise dispose of or hedge, directly or indirectly, any shares of our capital stock or
any securities convertible into or exercisable or exchangeable for shares of capital stock, for a period of ninety (90) days, after
the date of this prospectus, without the prior written consent of the representative. In connection with our IPO, our officers and
directors agreed not to sell, offer, agree to sell, contract to sell, hypothecate, pledge, grant any option to purchase, make any
short sale of, or otherwise dispose of or hedge, directly or indirectly, any shares of our capital stock or any securities convertible
into or exercisable or exchangeable until June 15, 2024, without the prior written consent of the representative. See the section
of this prospectus entitled “Underwriting” for additional information. |
The
number of shares of our common stock to be outstanding after this offering is based on 12,097,643 shares of our common stock outstanding
as of January 12, 2024, and excludes:
| |
● |
1,288,255
shares of our common stock issuable upon exercise
of outstanding options, with a weighted average exercise price of $1.36 per share, granted pursuant to our 2016 Stock Incentive
Plan, or the 2016 Plan, and our 2023 Stock Incentive Plan, or the 2023 Plan; |
| |
● |
approximately
323,736 shares of our common stock issuable upon exercise of outstanding warrants, with a weighted average exercise price of $4.75
per share; |
| |
● |
up
to 450,000 shares of our common stock (and/or Pre-Funded Warrants to purchase up to 450,000 shares of our common stock
in lieu thereof) issuable pursuant to the underwriters’ over-allotment option; |
| |
● |
120,000 shares
issuable upon exercise of a warrant to be issued to the underwriter as part of its compensation in connection with this offering
(up to 138,000 shares if the over-allotment option is exercised in full) at an exercise price of $2.56 per share (125%
of the assumed public offering price per share of the shares of common stock sold in this offering); and |
| |
● |
242,345
shares of our common stock reserved for future grants under our 2016 Plan and 1,960,000 shares of our common stock reserved for future
grants under our 2023 Plan. |
Unless
we indicate otherwise or unless the context otherwise requires, all information in this prospectus assumes the following:
| |
● |
no
exercise of outstanding warrants or options described above; |
| |
● |
no exercise of the Pre-Funded Warrants; and |
| |
● |
no
exercise of the underwriters’ over-allotment option. |
SUMMARY
RISK FACTORS
Our
business is subject to numerous risks, including risks that may prevent us from achieving our business objectives or adversely affect
our business, results of operations, cash flows, and prospects. You should carefully consider the risks discussed below and further discussed
in the section “Risk Factors” immediately following this prospectus summary before investing in our common stock or
Pre-Funded Warrants.
| |
● |
We
are an early-stage clinical biopharmaceutical company with limited operating history; |
| |
● |
We
have a history of significant operating losses and anticipate continued operating losses for the foreseeable future; |
| |
● |
We
expect we will need additional financing to execute our business plan and fund operations, which additional financing may not be
available on reasonable terms, or at all; |
| |
● |
The
clinical and commercial utility of our microbial library and genetic engineering platform is uncertain and may never be realized; |
| |
● |
Our
product candidates are in early stages of development, and therefore they will require extensive additional preclinical and clinical
testing; |
| |
● |
We
will need to grow the size of our organization, and we may experience difficulties in managing this growth; |
| |
● |
We
currently have no sales and marketing organization; |
| |
● |
We
will be completely dependent for the foreseeable future on third parties to manufacture our product candidates for commercial sale; |
| |
● |
Our
business model includes the potential out-licensing of strains from our proprietary microbial library or our product candidates to
other pharmaceutical companies; however, technology licensing in the pharmaceutical industry is a lengthy process and subject to
several risks and factors outside of our control; |
| |
● |
Our
business may suffer with the loss of key personnel; |
| |
● |
If
product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization
of our product candidates; |
| |
● |
Our
business operations could suffer in the event of information technology systems’ failures or security breaches; |
| |
● |
We
face significant competition from other biotechnology and pharmaceutical companies targeting medical dermatological indications; |
| |
● |
Our
success is entirely dependent on our ability to obtain the marketing approval for our product candidates by the FDA and the regulatory
authorities in foreign jurisdictions in which we intend to market our product candidates, of which there can be no assurance; |
| |
● |
Our
clinical trials may fail to demonstrate substantial evidence of the safety and efficacy of our product candidates or any future product
candidates; |
| |
● |
Results
of preclinical studies of our product candidates may not be predictive of the results of future preclinical studies or clinical trials; |
| |
● |
Even
if we receive regulatory approval for any of our product candidates, we may not be able to successfully commercialize the product
and the revenue that we generate from its sales, if any, may be limited; |
| |
● |
Current
and future legislation may increase the difficulty and cost for us to obtain marketing approval of and commercialize our product
candidates and affect the prices we may obtain; |
| |
● |
It
is difficult and costly to protect our intellectual property rights, and we cannot ensure the protection of these rights; |
| |
● |
Our
product candidates may infringe the intellectual property rights of others, which could increase our costs and delay or prevent our
development and commercialization efforts; |
| |
● |
An
active, liquid and orderly trading market for our shares may not develop; |
| |
● |
Future
capital raises may dilute your ownership and have other adverse effects on our operations; |
| |
● |
The
market price of our shares may be subject to fluctuation and volatility; |
| |
● |
If
we fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately report our
financial results or prevent fraud; |
| |
● |
There is no market for Pre-Funded Warrants and one is
not expected to develop; |
| |
● |
Holders of the Pre-Funded Warrants purchased in this
offering will have no rights as common stockholders until such holders exercise such warrants and acquire our common stock; |
| |
● |
We
ratified certain corporate actions pursuant to Section 204 of the Delaware General Corporate Law, or DGCL; however, there can be
no assurance that claims will not be made to challenge the validity of the ratification or the related corporate actions; and |
| |
● |
Our
charter documents and Delaware law may inhibit a takeover that stockholders consider favorable. |
SUMMARY
FINANCIAL DATA
The
following tables summarize our financial data. You should read this summary financial data together with the section entitled “Management’s
Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and related notes that
are included elsewhere in this prospectus. The financial information as of and for the fiscal years ended December 31, 2022 and 2021
is derived from the audited financial statements that are included elsewhere in this prospectus. The financial information as of and
for the nine months ended September 30, 2023 and 2022 are derived from our unaudited financial statements included elsewhere in this
prospectus. The unaudited financial statements were prepared on the same basis as the audited financial statements. Our management believes
that the unaudited financial statements reflect all adjustments necessary for the fair presentation of the financial condition and results
of operations for such periods. Our historical results are not necessarily indicative of the results that may be expected in the future.
| | |
Nine
Months Ended
September
30, | | |
Years
Ended
December 31, | |
| (in
thousands, except share amounts) | |
2023 | | |
2022 | | |
2022 | | |
2021 | |
| Statement of Operations Data | |
(unaudited) | | |
(unaudited) | | |
| | |
| |
| Revenues | |
$ | 596 | | |
$ | 254 | | |
$ | 284 | | |
$ | 110 | |
| Net loss | |
$ | (8,830 | ) | |
$ | (6,634 | ) | |
$ | (10,680 | ) | |
$ | (8,940 | ) |
| Net loss per share, basic and diluted | |
$ | (1.97 | ) | |
| (8.25 | ) | |
$ | (12.74 | ) | |
$ | (11.20 | ) |
| | |
September
30, 2023 | |
| (in
thousands) | |
Actual | | |
As
Adjusted(1) | |
| Balance Sheet Data: | |
(unaudited) | | |
(unaudited) | |
| Cash and cash equivalents | |
$ | 4,400 | | |
$ | 9,527 | |
| Working capital | |
$ | 3,410 | | |
$ | 8,537 | |
| Total assets | |
$ | 7,490 | | |
$ | 12,617 | |
| Additional paid-in capital | |
$ | 51,475 | | |
$ | 56,601 | |
| Total stockholders’
equity | |
$ | 5,332 | | |
$ | 10,459 | |
| (1) | The
as adjusted column gives effect to the sale by us of 3,000,000 shares of common stock
(and/or Pre-Funded Warrants to purchase up to 3,000,000 shares of our common stock in
lieu thereof) offered by this prospectus at the assumed public offering price
of $2.05 per share, the last reported sale price of our common stock on The NYSE
American on January 18, 2024, after deducting underwriting discounts and commissions
and estimated offering costs payable by us. |
RISK
FACTORS
Any
investment in our common stock involves a high degree of risk. You should carefully consider the risks described below, which we believe
represent certain of the material risks to our business, together with the information contained elsewhere in this prospectus, before
you make a decision to invest in our common stock. Please note that the risks highlighted here are not the only ones that we may face.
For example, additional risks presently unknown to us or that we currently consider immaterial or unlikely to occur could also impair
our operations. If any of the following events occur or any additional risks presently unknown to us actually occur, our business, financial
condition and operating results may be materially adversely affected. In that event, the trading price of our common stock could decline
and you could lose all or part of your investment.
Risks
Relating to Our Business
We
are an early-stage clinical biopharmaceutical company with limited operating history.
We
are an early-stage clinical biopharmaceutical company incorporated on January 2, 2014, and have limited operating history. We have not
commenced revenue-producing operations apart from limited grant and service revenue. To date, our operations have consisted of the development
of our proprietary microbial library, the identification, characterization, genetic engineering and testing of certain bacterial species
to provide therapeutic effect and the development of our initial product candidates. Our limited operating history makes it difficult
for potential investors to evaluate our technology or prospective operations. As an early-stage clinical biopharmaceutical company, we
are subject to all the risks inherent in the organization, financing, expenditures, complications and delays involved with a new business.
Accordingly, you should consider our prospects in light of the costs, uncertainties, delays and difficulties frequently encountered by
companies in the early stages of development, especially early-stage clinical-stage biopharmaceutical companies such as ours. Potential
investors should carefully consider the risks and uncertainties that a company with a limited operating history will face. In particular,
potential investors should consider that we may be unable to:
| |
● |
successfully
implement or execute our business plan, or that our business plan is sound; |
| |
● |
successfully
complete preclinical and clinical trials and obtain regulatory approval for the marketing of our product candidates; |
| |
● |
successfully
demonstrate a favorable differentiation between our precision dermatological product candidates and the current products on the market; |
| |
● |
successfully
contract for the manufacture of our clinical drug products and establish a commercial drug supply; |
| |
● |
secure
market exclusivity or adequate intellectual property protection for our product candidates; |
| |
● |
attract
and retain an experienced management and advisory team; and |
| |
● |
raise
sufficient funds in the capital markets to effectuate our business plan, including product and clinical development, regulatory approval
and commercialization for our product candidates. |
Investors
should evaluate an investment in us in light of the uncertainties encountered by developing companies in a competitive environment. There
can be no assurance that our efforts will be successful or that we will ultimately be able to attain profitability. If we cannot successfully
execute any one of the foregoing, our business may not succeed and your investment will be adversely affected. You must be prepared to
lose all of your investment.
We
have a history of significant operating losses and anticipate continued operating losses for the foreseeable future.
For
the fiscal years ended December 31, 2022 and 2021, we incurred a net loss of $10.7 million and $8.9 million, respectively, and $8.8 million
and $6.6 million for the nine months ended September 30, 2023 and 2022, respectively. As of September 30, 2023, we had an accumulated
deficit of $46.1 million. Following completion of this offering, we expect to continue to incur substantial expenses without any meaningful
revenues unless and until we are able to obtain regulatory approval and successfully commercialize at least one of our product candidates.
We also believe that, at a minimum, it will take us four to six years from the closing of the offering for us to obtain regulatory approval
of our first drug candidates, assuming we are able to get regulatory approval at all. Even if we are able to commercialize our product
candidates, there can be no assurance that we will generate significant revenues or ever achieve profitability.
We
expect to have significant research, regulatory and development expenses as we advance our product candidates towards commercialization.
As a result, we expect to incur substantial losses for the foreseeable future, and these losses will be increasing. We are uncertain
when or if we will be able to achieve or sustain profitability. If we achieve profitability in the future, we may not be able to sustain
profitability in subsequent periods. Failure to become and remain profitable may impair our ability to sustain operations and adversely
affect our business and our ability to raise capital. If we are unable to generate positive cash flow within a reasonable period of time,
we may be unable to further pursue our business plan or continue operations, in which case you may lose your entire investment.
The
report of our independent registered public accounting firm for the year ended December 31, 2022 states that due to our accumulated deficit,
recurring and negative cash flow from operations there is substantial doubt about our ability to continue as a going concern.
We
expect we will need additional financing to execute our business plan and fund operations, which additional financing may not be available
on reasonable terms or at all.
As
of September 30, 2023, we had total assets of $7.5 million and working capital of $3.4 million. We believe that net proceeds of this
offering, along with our cash on hand as of the date of this prospectus, may not be sufficient to cover our proposed plan of operations
over, at least, the next 12 months. We intend
to seek additional funds through various financing sources, including the sale of our equity, licensing fees for our technology and joint
ventures with industry partners. In addition, we will consider alternatives to our current business plan that may enable to us to achieve
revenue producing operations and meaningful commercial success with a smaller amount of capital. However, there can be no guarantees
that such funds will be available on commercially reasonable terms, if at all. If such financing is not available on satisfactory terms,
we may be unable to further pursue our business plan and we may be unable to continue operations, in which case you may lose your entire
investment.
The
clinical and commercial utility of our microbial library and genetic engineering platform is uncertain and may never be realized.
We
have built a proprietary platform that includes a microbial library comprised of approximately 1,500 unique bacterial strains that can
be screened for unique therapeutic characteristics. The platform is augmented by artificial intelligence, machine learning and genetic
engineering technologies. To date, our focus is on the development of genetically engineered strains of S. epidermidis, which
we consider to be an optimal therapeutic candidate species for engineering of dermatologic therapies. However, we believe that the genetic
engineering of S. epidermidis is a novel and unproven mode of therapy. We recently initiated our Phase 1b clinical trial for ATR-12
and expect to enroll our first patient in the first quarter of 2024, and expect to file an IND for a Phase 1b clinical trial of ATR-04
by the first half of 2024. However, as of the date of this prospectus, we have tested and evaluated our proprietary strains of S.
epidermidis in preclinical studies and have not conducted any clinical trials designed to evaluate safety, tolerability or efficacy.
Furthermore, success in early clinical trials does not ensure that large-scale clinical trials will be successful, nor does it predict
final results. Even after the completion of our proposed Phase 1b clinical trials, our initial product candidates will have only been
tested in a small number of patients. Results from these clinical trials may not necessarily be indicative of the safety and tolerability
or efficacy of our product candidates or our as we expand into larger clinical trials. Until such time, if ever, as we are able to provide
the FDA with substantial clinical evidence to support a claim of safety, efficacy, purity and potency sufficient to enable the FDA to
approve our proprietary product candidates for any indication, our proprietary microbial library and genetic engineering platform will
remain unproven.
Our
product candidates are in early-stage clinical trials or early stages of preclinical development, and therefore they will require extensive
additional preclinical and clinical testing. Success in preclinical studies or early-stage clinical trials may not be indicative of results
in future clinical trials and we cannot assure you that any ongoing, planned or future clinical trials will lead to results sufficient
for the necessary regulatory approvals.
Because
our product candidates are in early stages clinical trials or of preclinical development, they will require extensive preclinical and
clinical testing. We recently initiated our Phase 1b clinical trial for ATR-12 and expect to enroll our first patient in the first quarter
of 2024, and expect to file an IND for a Phase 1b clinical trial of ATR-04 by the first half of 2024. However, we have not conducted
meaningful preclinical studies for any of our other product candidates. Success in preclinical testing and early-stage clinical trials
does not ensure that later clinical trials will generate the same results or otherwise provide adequate data to demonstrate the efficacy
and safety of a product candidate. Preclinical studies and Phase 1b clinical trials are primarily designed to test safety, to study pharmacokinetics
and pharmacodynamics and to understand the side effects of product candidates at various doses and schedules. Phase 1b clinical trials
also test how well a certain disease responds to a new treatment. Success in preclinical studies and earlier clinical trials does not
ensure that later efficacy trials will be successful, nor does it predict final results. Our product candidates may fail to show the
desired safety and efficacy in clinical development despite positive results in preclinical studies or even if they successfully advance
through earlier clinical trials.
In
addition, the design of a clinical trial can determine whether its results will support approval of a product, and flaws in the design
of a clinical trial may not become apparent until the clinical trial is well advanced. As an organization, we have limited experience
designing clinical trials and may be unable to design and execute a clinical trial to support regulatory approval. Many companies in
the pharmaceutical and biotechnology industries have suffered significant setbacks, including failure in late-stage clinical trials even
after achieving promising results in preclinical testing and earlier clinical trials. Data obtained from preclinical and clinical activities
are subject to varying interpretations, which may delay, limit or prevent regulatory approval.
Further,
we cannot predict with any certainty if or when we might submit a Biologics License Application, or BLA, for regulatory approval for
any of our product candidates or whether any such BLA will be accepted for review by the FDA, or whether any BLA will be approved upon
review. Even if our clinical trials are completed as planned, we cannot be certain that their results will support our proposed indications.
Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and we cannot
be sure that the results of later clinical trials will replicate the results of prior clinical trials and preclinical testing. The clinical
trial process may fail to demonstrate that our product candidates are safe and effective for their proposed uses. This failure could
cause us to abandon a product candidate and may delay development of other product candidates. Any delay in, or termination of, our clinical
trials will delay and possibly preclude the filing of any BLAs with the FDA and, ultimately, our ability to commercialize our product
candidates and generate product revenues.
We
will need to grow the size of our organization, and we may experience difficulties in managing this growth.
As
our development and commercialization plans and strategies develop, we will need to expand the size of our employee and consultant/contractor
base. Future growth would impose significant added responsibilities on members of management, including the need to identify, recruit,
maintain, motivate and integrate additional employees. In addition, our management may have to divert a disproportionate amount of its
attention away from our day-to-day activities and devote a substantial amount of time to managing these growth activities. Our future
financial performance and our ability to commercialize our product candidates and any other future product candidates and our ability
to compete effectively will depend, in part, on our ability to effectively manage our future growth.
If
we are not successful in attracting and retaining highly qualified personnel, we may not be able to successfully implement our business
strategy. In addition, the loss of the services of our senior management would adversely impact our business prospects.
Our
management team has expertise in many different aspects of drug development and commercialization. However, our ability to compete in
the highly competitive pharmaceuticals industry depends in large part upon our ability to attract and retain highly qualified managerial,
scientific and medical personnel. We will need to hire additional personnel as we further develop our product candidates. Competition
for skilled personnel in our market is intense and competition for experienced scientists may limit our ability to hire and retain highly
qualified personnel on acceptable terms. Despite our efforts to retain valuable employees, members of our management, scientific and
medical teams may terminate their employment with us on short notice. The loss of the services of any of our executive officers or other
key employees, or our inability to hire targeted executives, could potentially harm our business, operating results or financial condition.
In particular, we believe that the loss of the services of our chief executive officer would have a material adverse effect on our business.
Other
biopharmaceutical companies with which we compete for qualified personnel have greater financial and other resources, different risk
profiles, and a longer history in the industry than we do. They also may provide more diverse opportunities and better chances for career
advancement. Some of these characteristics may be more appealing to high-quality candidates than what we have to offer. If we are unable
to continue to attract and retain high-quality personnel, the rate and success at which we can develop and commercialize product candidates
would be limited.
We
currently have no sales and marketing organization. If we are unable to establish satisfactory sales and marketing capabilities or secure
a third-party sales and marketing relationship, we may not be able to successfully commercialize any of our product candidates.
At
present, we have no sales or marketing personnel. Upon and subject to initial receipt of the requisite regulatory approvals for one or
more of our drug products, we plan to build focused capabilities in the United States to commercialize our development programs focused
on live biotherapeutic products and recombinant proteins for the treatment of skin diseases, where we believe the patient populations
and medical specialists for the indications we are targeting are sufficiently concentrated to allow us to effectively promote our products
with a targeted sales team. In other markets for which commercialization may be less capital efficient for us, we may selectively pursue
strategic collaborations with third parties in order to maximize the commercial potential of our product candidates. In some cases, we
may pursue the licensing of our microbial library or patent rights or enter into a joint development arrangement. If we are not successful
in recruiting sales and marketing personnel and building a sales and marketing infrastructure or entering into appropriate collaboration
arrangements with third parties, we will have difficulty successfully commercializing our product candidates, which would adversely affect
our business, operating results and financial condition.
Even
if we enter into third-party marketing and distribution arrangements, we may have limited or no control over the sales, marketing and
distribution activities of these third parties. Our future revenues may depend heavily on the success of the efforts of these third parties.
In terms of establishing a sales and marketing infrastructure, we will have to compete with established and well-funded pharmaceutical
and biotechnology companies to recruit, hire, train and retain sales and marketing personnel. Factors that may inhibit our efforts to
build an internal sales organization or enter into collaboration arrangements with third parties include:
| |
● |
our
inability to recruit and retain adequate numbers of effective sales and marketing personnel; |
| |
● |
the
inability of sales personnel to obtain access to or persuade adequate numbers of physicians to prescribe any of our product candidates; |
| |
● |
the
lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies
with more extensive product lines; and |
| |
● |
unforeseen
costs and expenses associated with creating an internal sales and marketing organization. |
We
will be completely dependent for the foreseeable future on third parties to manufacture our product candidates for commercial sale, and
the commercialization of our product candidates could be halted, delayed or made less profitable if those third parties fail to obtain
manufacturing approval from the FDA or comparable foreign regulatory authorities, fail to provide us with sufficient quantities of our
product candidates or fail to do so at acceptable quality levels or prices.
We
do not own or operate manufacturing facilities for the commercial production of our current product candidates. We currently rely on
third-party contract manufacturers for all of our required raw materials, manufacturing devices, and active pharmaceutical ingredients
for our preclinical research and clinical trials. Although we are able to manufacture finished product in our Groton Connecticut facility
for our clinical trials, we will rely on third parties for the manufacture of our finished product for commercial sale. We do not have
long-term agreements with any of these third parties. We also do not have any current contractual relationship for commercial supplies.
We intend to enter into agreements with third-party contract manufacturers and one or more backup manufacturers for future production.
We are analyzing the feasibility of building manufacturing capabilities for future development and commercial quantities of any products
that we develop. Such products will need to be manufactured in facilities, and by processes, that comply with the requirements of the
FDA and the regulatory agencies of other jurisdictions in which we are seeking approval. In the meantime, we will be obligated to rely
on contract manufacturers for our preclinical research and clinical trials and commercial production, if and when any of our product
candidates are approved for commercialization.
The
facilities used by us or any contract manufacturer to manufacture our raw materials, manufacturing devices, active pharmaceutical ingredients
and finished products must be approved by the FDA or comparable foreign regulatory authorities. Such approvals are subject to inspections
that will be conducted after we submit a BLA to the FDA or their equivalents to other relevant regulatory authorities. Until such time,
if ever, as we establish our own manufacturing facilities, we will not control the manufacturing process of our product candidates, and
will be completely dependent on our contract manufacturing partners for compliance with Current Good Manufacturing Practices, or cGMPs,
for manufacture of our raw materials, manufacturing devices, active pharmaceutical ingredients and finished products. These cGMP regulations
cover all aspects of the manufacturing, testing, quality control, storage, distribution and record keeping relating to our product candidates.
If our contract manufacturers do not successfully manufacture material that conforms to our specifications and the strict regulatory
requirements of the FDA or others, we will not be able to secure or maintain regulatory approval for product made at their manufacturing
facilities. If the FDA or a comparable foreign regulatory authority does not approve these facilities for the manufacture of our product
candidates or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which could
significantly delay our clinical trials and impact our ability to develop, manufacture, obtain regulatory approval for or market our
product candidates, if approved. Likewise, we could be negatively impacted if any of our contract manufacturers elect to discontinue
their business relationship with us.
Our
contract manufacturers will be subject to ongoing periodic unannounced inspections by the FDA and corresponding state and foreign agencies
for compliance with cGMPs and similar regulatory requirements. We will not have control over our contract manufacturers’ compliance
with these regulations and standards. Failure by any of our contract manufacturers to comply with applicable regulations could result
in sanctions being imposed on us, including fines, injunctions, civil penalties, failure to grant approval to market any of our product
candidates, delays, suspensions or withdrawals of approvals, inability to supply product, operating restrictions and criminal prosecutions,
any of which could significantly and adversely affect our business. In addition, we will not have control over the ability of our contract
manufacturers to maintain adequate quality control, quality assurance and qualified personnel. Failure by our contract manufacturers
to comply with or maintain any of these standards could adversely affect our ability to develop, manufacture, obtain regulatory approval
for or market any of our product candidates, if approved.
If,
for any reason, these third parties are unable or unwilling to perform we may not be able to locate alternative manufacturers or formulators
or enter into favorable agreements with them and we cannot be certain that any such third parties will have the manufacturing capacity
to meet future requirements. If these manufacturers or any alternate manufacturer of finished drug product experiences any significant
difficulties in its respective manufacturing processes for our required raw materials, manufacturing devices, active pharmaceutical ingredients
or finished product or should cease doing business with us for any reason, we could experience significant delays in our clinical trials
and significant interruptions in the supply of any of our product candidates or may not be able to create a supply of our product candidates
at all.
Any
manufacturing problem or the loss of a contract manufacturer could be disruptive to our operations and result in development and clinical
trial delays and lost sales. Additionally, we will rely on third parties to supply the raw materials needed to manufacture our product
candidates. Any such reliance on suppliers may involve several risks, including a potential inability to obtain critical materials and
reduced control over production costs, delivery schedules, reliability and quality. Any unanticipated disruption to the operation of
one of our contract manufacturers caused by problems with suppliers could delay our shipment of any of our product candidates, increase
our cost of goods sold and result in delays in clinical trials or lost sales.
Our
business model includes the potential out-licensing of strains from our proprietary microbial library or our product candidates to other
biopharmaceutical companies, however technology licensing in the biopharmaceutical industry is a lengthy process and subject to several
risks and factors outside of our control, and we cannot forecast our ability to successfully out-license our technology or the length
of time it takes to establish a new licensing relationship.
Our
business model includes the potential out-licensing or joint development of strains from our proprietary microbial library or our product
candidates to other biopharmaceutical companies. Any such arrangement would typically begin with preliminary feasibility testing and
evaluation by our potential partner or licensee. Assuming the feasibility testing is successful, our ability to convert the successful
test into a commercial license or joint development agreement is dependent on a number of risks and factors, many of which are outside
our control, including:
| |
● |
the
rate of adoption and incorporation of new technologies, by members of the pharmaceutical industry generally; |
| |
● |
our
potential licensee’s internal evaluation of the economic benefits of marketing a dermatological product that may be competitive
with other products currently in development or commercial sale by our potential partner or licensee regardless of the perceived
benefits or advantages of our technology or product; |
| |
● |
our
potential partner’s/licensee’s internal budgetary and product development issues, including their ability to commit the
capital and human resources towards the development and commercialization of our technology or product; and |
| |
● |
our
potential partner’s/licensee’s willingness to accept our requirements for upfront fees and ongoing royalties. |
In
addition, we believe that in many cases our potential partners or licensee may engage with us in the early-stage feasibility testing
as part of their evaluation of multiple drug and drug delivery options and prior to making any decision or commitment to the development
of a new drug product. Consequently, even if our platform is successful in early feasibility studies, our potential partner/licensee
may decide, for reasons unrelated to the performance of our technology, not to enter into a license agreement with us. Therefore, we
are unable to predict the degree to which our proposed licensing model will be successful.
If
product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization
of our product candidates.
We
will face a potential risk of product liability as a result of the clinical testing of our product candidates and will face an even greater
risk of such liability if we commercialize any of our product candidates. For example, we may be sued if any product we develop, including
any of our product candidates, or any materials that we use in our product candidates allegedly causes injury or is found to be otherwise
unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects
in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability and a breach
of warranties. In the U.S., claims could also be asserted against us under state consumer protection acts. If we cannot successfully
defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of
our product candidates. Even successful defense of these claims would require us to employ significant financial and management resources.
Regardless of the merits or eventual outcome, liability claims may result in:
| |
● |
decreased
demand for any of our product candidates or any future products that we may develop; |
| |
● |
injury
to our reputation; |
| |
● |
failure
to obtain regulatory approval for our product candidates; |
| |
● |
withdrawal
of participants in our clinical trials; |
| |
● |
costs
associated with our defense of the related litigation; |
| |
● |
a
diversion of our management’s time and our resources; |
| |
● |
substantial
monetary awards to trial participants or patients; |
| |
● |
product
recalls, withdrawals or labeling, marketing or promotional restrictions; |
| |
● |
the
inability to commercialize some or all of our product candidates; and |
| |
● |
a
decline in the value of our stock. |
As
of the date of this prospectus, we carry product liability insurance that we consider adequate for our current level of clinical testing
and development. However, we will need additional product liability coverage at the time we commence commercial sale of our initial product.
Our inability to obtain and retain sufficient product liability insurance at an acceptable cost to protect against potential product
liability claims could prevent or inhibit the commercialization of products we develop. Although we will endeavor to obtain and maintain
such insurance in coverage amounts we deem adequate, any claim that may be brought against us could result in a court judgment or settlement
in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage.
Our insurance policies would also have various exclusions, and we may be subject to a product liability claim for which we have no coverage.
As a result, we may have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or
that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts.
Our
business operations could suffer in the event of information technology systems’ failures or security breaches.
While
we believe that we have implemented adequate security measures within our internal information technology and networking systems, our
information technology systems may be subject to security breaches, damages from computer viruses, natural disasters, terrorism, and
telecommunication failures. Any system failure or security breach could cause interruptions in our operations in addition to the possibility
of losing proprietary information and trade secrets. To the extent that any disruption or security breach results in inappropriate disclosure
of our confidential information, our competitive position may be adversely affected and we may incur liability or additional costs to
remedy the damages caused by these disruptions or security breaches.
We
face significant competition from other biotechnology and pharmaceutical companies targeting medical dermatological indications, and
our operating results will suffer if we fail to compete effectively.
The
dermatological therapies market is highly competitive and led by significant technologic developments. We anticipate that, if we are
successful in obtaining regulatory approval of our candidates, we will face significant competition from other approved therapies or
drugs that will become available in our industry. Even if another branded, generic, or OTC product is less effective, it may be quickly
adopted by physicians and patients than our product based upon cost or convenience.
Risks
Related to Product Regulation
Our
success is entirely dependent on our ability to obtain the marketing approval for our product candidates by the FDA and the regulatory
authorities in foreign jurisdictions in which we intend to market our product candidates, of which there can be no assurance.
We
are not permitted to market our product candidates as prescription pharmaceutical products in the United States until we receive approval
of a BLA from the FDA, or in any foreign countries until we receive the requisite approval from such countries. In the United States,
the FDA generally requires the completion of clinical trials of each biologic to establish its safety and efficacy and extensive pharmaceutical
development to ensure its quality before a BLA is approved. Of the large number of biologics in development, only a small percentage
result in the submission of a BLA to the FDA and even fewer are eventually approved for commercialization. As of the date of this prospectus,
we have not submitted a BLA to the FDA or comparable applications to other regulatory authorities for any of our product candidates.
Our
success depends on our receipt of the regulatory approvals described above, and the issuance of such regulatory approvals is uncertain
and subject to a number of risks, including the following:
| |
● |
such
authorities may disagree with the number, design, size, conduct or implementation of our clinical trials or any of our collaborators’
clinical trials; |
| |
● |
such
authorities may disagree with our interpretation of data from preclinical studies or clinical trials or the use of results from studies
that served as precursors to our current or future product candidates; |
| |
● |
the
results of toxicology studies may not support the filing of an Investigational New Drug Application, or IND, or a BLA for our product
candidates; |
| |
● |
the
FDA or comparable foreign regulatory authorities or Institutional Review Boards, or IRBs, may disagree with the design or implementation
of our clinical trials; |
| |
● |
we
may not be able to provide acceptable evidence of our product candidates’ safety and efficacy; |
| |
● |
the
results of our clinical trials may not be satisfactory or may not meet the level of statistical or clinical significance required
by the FDA, European Medicines Agency, or EMA, or other regulatory agencies for us to receive marketing approval for any of our product
candidates; |
| |
● |
the
dosing of our product candidates in a particular clinical trial may not be at an optimal level; |
| |
● |
patients
in our clinical trials may suffer adverse effects for reasons that may or may not be related to our product candidates; |
| |
● |
the
data collected from clinical trials may not be sufficient to support the submission of a BLA or other submission or to obtain regulatory
approval in the United States or elsewhere; |
| |
● |
the
FDA may require development of a Risk Evaluation and Mitigation Strategy, or REMS, as a condition of approval; |
| |
● |
the
FDA or comparable foreign regulatory authorities may fail to approve the manufacturing processes or facilities of third-party manufacturers
with which we contract for clinical and commercial supplies; and |
| |
● |
the
approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering
our clinical data insufficient for approval of our product candidates. |
The
process of obtaining regulatory approvals is expensive, often takes many years, if approval is obtained at all, and can vary substantially
based upon, among other things, the type, complexity and novelty of the product candidates involved, the jurisdiction in which regulatory
approval is sought and the substantial discretion of the regulatory authorities. Changes in regulatory approval policies during the development
period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for a submitted product application
may cause delays in the approval or rejection of an application. Regulatory approval obtained in one jurisdiction does not necessarily
mean that a product candidate will receive regulatory approval in all jurisdictions in which we may seek approval, but the failure to
obtain approval in one jurisdiction may negatively impact our ability to seek approval in a different jurisdiction. Failure to obtain
regulatory approval for our product candidates for the foregoing, or any other reasons, will prevent us from commercializing our product
candidates, and our ability to generate revenue will be materially impaired.
In
addition, the FDA, EMA or other regulatory agencies may also approve a product candidate for fewer or more limited indications than we
request, may impose significant limitations related to use restrictions for certain age groups, warnings, precautions or contraindications
or may grant approval contingent on the performance of costly post-marketing clinical trials or risk mitigation requirements. The FDA,
EMA or other regulatory agencies may also not accept the labeling claims that we believe would be necessary or desirable for the successful
commercialization of our product candidates.
In
December 2022, the U.S. Congress enacted a new law, the Modernization of Cosmetics Regulation Act of 2022, or MOCRA. MOCRA will require
a cosmetic manufacturer or importer to: ensure that it has on hand substantiation of the safety of its products and ingredients; meet
increased registration, record-keeping and reporting requirements; include fragrance and allergen information on its labeling; and be
prepared to meet FDA’s to be promulgated good manufacturing practices requirements. These additional requirements may impact budgets
and timelines.
Our
clinical trials may fail to demonstrate substantial evidence of the safety and efficacy of our product candidates or any future product
candidates, which would prevent or delay or limit the scope of regulatory approval and commercialization.
Our
business model depends entirely on the successful development, regulatory approval and commercialization of our product candidates, which
may never occur. We recently initiated our Phase 1b clinical trial for ATR-12 and expect to enroll our first patient in the first quarter
of 2024, and expect to file an IND for a Phase 1b clinical trial of ATR-04 by the first half of 2024. However, all of our other product
candidates are in the early stages of development and as of the date of this prospectus we have not progressed any of our product candidates,
other than ATR-12 and ATR-04, beyond performance characterization and animal testing We may not be successful in obtaining approval from
the FDA or comparable foreign regulatory authorities to start clinical trials for ATR-04 or any of our other product candidates. If we
do not obtain such approvals as presently planned, the time in which we expect to commence clinical programs for any product candidate
will be extended and such extension will increase our expenses, delay our potential receipt of any revenues and increase our need for
additional capital. Moreover, there is no guarantee that we will receive approval to commence human clinical trials or, if we do receive
approval, that our clinical trials will be successful or that we will continue clinical development in support of an approval from the
FDA or comparable foreign regulatory authorities for any indication. We note that most product candidates never reach the clinical development
stage and even those that do commence clinical development have only a small chance of successfully completing clinical development and
gaining regulatory approval. Success in early phases of preclinical and clinical trials does not ensure that later clinical trials will
be successful, and interim results of a clinical trial do not necessarily predict final results. A failure of one or more of our clinical
trials can occur at any stage of testing. We may experience numerous unforeseen events during, or as a result of, the clinical trial
process that could delay or prevent our ability to receive regulatory approval or commercialize our product candidates. Therefore, our
business currently depends entirely on the successful development, regulatory approval and commercialization of our product candidates,
which may never occur.
Results
of preclinical studies of our product candidates may not be predictive of the results of future preclinical studies or clinical trials.
To
obtain the requisite regulatory approvals to market and sell any of our product candidates, we must demonstrate through extensive preclinical
studies and clinical trials that our product candidates are safe, pure, and potent in humans. Before an IND can be submitted to the FDA
and become effective, which is a prerequisite for conducting clinical trials on human subjects in the United States, a product candidate
must successfully progress through extensive preclinical studies, which include preclinical laboratory testing, animal studies, and formulation
studies in accordance with Good Laboratory Practices. Success in preclinical studies does not ensure that later preclinical studies or
clinical trials will be successful. A number of companies in the biotechnology and pharmaceutical industries have suffered significant
setbacks in clinical trials, even after positive results in earlier preclinical studies. These setbacks have been caused by, among other
things, preclinical findings made while clinical trials were underway and safety or efficacy observations made in clinical trials, including
previously unreported adverse events. The design of a clinical trial can determine whether its results will support approval of a product,
and flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced. Further, we or our investigators
may have little control over whether subjects comply with important aspects of clinical trial protocols. In addition, preclinical and
clinical data are often susceptible to varying interpretations and analyses. Notwithstanding any potential promising results in earlier
studies, we cannot be certain that we will not face similar setbacks. In addition, the results of our preclinical animal studies may
not be predictive of the results of outcomes in subsequent clinical trials on human subjects. Product candidates in clinical trials may
fail to show the desired pharmacological properties or safety and efficacy traits despite having progressed through preclinical studies.
If
we fail to receive positive results in preclinical studies or clinical trials of our product candidates, the development timeline and
regulatory approval and commercialization prospects for our most advanced product candidates, and, correspondingly, our business and
financial prospects would be negatively impacted.
Any
termination or suspension of, or delays in the commencement or completion of, any necessary studies of any of our product candidates
for any indications could result in increased costs to us, delay or limit our ability to generate revenue and adversely affect our commercial
prospects.
The
commencement and completion of clinical studies can be delayed for a number of reasons, including delays related to:
| |
● |
the
FDA or a comparable foreign regulatory authority failing to grant permission to proceed and placing the clinical study on hold; |
| |
● |
subjects
for clinical testing failing to enroll or remain enrolled in our trials at the rate we expect; |
| |
● |
a
facility manufacturing any of our product candidates being ordered by the FDA or other government or regulatory authorities to temporarily
or permanently shut down due to violations of cGMP requirements or other applicable requirements, or cross-contaminations of product
candidates in the manufacturing process; |
| |
● |
any
changes to our manufacturing process that may be necessary or desired; |
| |
● |
subjects
choosing an alternative treatment for the indications for which we are developing our product candidates, or participating in competing
clinical studies; |
| |
● |
subjects
experiencing severe or unexpected drug-related adverse effects; |
| |
● |
reports
from clinical testing on similar technologies and products raising safety and/or efficacy concerns; |
| |
● |
third-party
clinical investigators losing their license or permits necessary to perform our clinical trials, not performing our clinical trials
on our anticipated schedule or employing methods consistent with the clinical trial protocol, cGMP requirements, or other third parties
not performing data collection and analysis in a timely or accurate manner; |
| |
● |
inspections
of clinical study sites by the FDA, comparable foreign regulatory authorities, or IRBs finding regulatory violations that require
us to undertake corrective action, result in suspension or termination of one or more sites or the imposition of a clinical hold
on the entire study, or that prohibit us from using some or all of the data in support of our marketing applications; |
| |
● |
third-party
contractors becoming debarred or suspended or otherwise penalized by the FDA or other government or regulatory authorities for violations
of regulatory requirements, in which case we may need to find a substitute contractor, and we may not be able to use some or any
of the data produced by such contractors in support of our marketing applications; |
| |
● |
one
or more IRBs refusing to approve, suspending or terminating the study at an investigational site, precluding enrollment of additional
subjects, or withdrawing its approval of the trial; reaching agreement on acceptable terms with prospective contract research organizations,
or CROs, and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different
CROs and trial sites; |
| |
● |
deviations
of the clinical sites from trial protocols or dropping out of a trial; |
| |
● |
adding
new clinical trial sites; |
| |
● |
the
inability of the CRO to execute any clinical trials for any reason; and |
| |
● |
government
or regulatory delays or “clinical holds” requiring suspension or termination of a trial. |
Product
development costs for any of our product candidates will increase if we have delays in testing or approval or if we need to perform more
or larger clinical studies than planned. Additionally, changes in regulatory requirements and policies may occur and we may need to amend
study protocols to reflect these changes. Amendments may require us to resubmit our study protocols to the FDA, comparable foreign regulatory
authorities, and IRBs for reexamination, which may impact the costs, timing or successful completion of that study. If we experience
delays in completion of, or if we, the FDA or other regulatory authorities, the IRB, or other reviewing entities, or any of our clinical
study sites suspend or terminate any of our clinical studies of any of our product candidates, its commercial prospects may be materially
harmed and our ability to generate product revenues will be delayed. Any delays in completing our clinical trials will increase our costs,
slow down our development and approval process and jeopardize our ability to commence product sales and generate revenues. Any of these
occurrences may harm our business, financial condition and prospects significantly. In addition, many of the factors that cause, or lead
to, termination or suspension of, or a delay in the commencement or completion of, clinical studies may also ultimately lead to the denial
of regulatory approval of our product candidates. In addition, if one or more clinical studies are delayed, our competitors may be able
to bring competing products to market before we do, and the commercial viability of any of our affected product candidates could be significantly
reduced.
Even
if we receive regulatory approval for any of our product candidates, we may not be able to successfully commercialize the product and
the revenue that we generate from its sales, if any, may be limited.
If
approved for marketing, the commercial success of our product candidates will depend upon each product’s acceptance by the medical
community, including physicians, patients and health care payors. The degree of market acceptance for any of our product candidates will
depend on a number of factors, including:
| |
● |
demonstration
of clinical safety and efficacy; |
| |
● |
relative
convenience, dosing burden and ease of administration; |
| |
● |
the
prevalence and severity of any adverse effects; |
| |
● |
the
willingness of physicians to prescribe our product candidates, and the target patient population to try new therapies; |
| |
● |
efficacy
of our product candidates compared to competing products; |
| |
● |
the
introduction of any new products that may in the future become available targeting indications for which our product candidates may
be approved; |
| |
● |
new
procedures or therapies that may reduce the incidences of any of the indications in which our product candidates may show utility; |
| |
● |
pricing
and cost-effectiveness; |
| |
● |
the
inclusion or omission of our product candidates in applicable therapeutic and vaccine guidelines; |
| |
● |
the
effectiveness of our own or any future collaborators’ sales and marketing strategies; |
| |
● |
limitations
or warnings contained in approved labeling from regulatory authorities; |
| |
● |
our
ability to obtain and maintain sufficient third-party coverage or reimbursement from government health care programs, including Medicare
and Medicaid, private health insurers and other third-party payors or to receive the necessary pricing approvals from government
bodies regulating the pricing and usage of therapeutics; and |
| |
● |
the
willingness of patients to pay out-of-pocket in the absence of third-party coverage or reimbursement or government pricing approvals. |
If
any of our product candidates are approved, but do not achieve an adequate level of acceptance by physicians, health care payors, and
patients, we may not generate sufficient revenue and we may not be able to achieve or sustain profitability. Our efforts to educate the
medical community and third-party payors on the benefits of our product candidates may require significant resources and may never be
successful.
In
addition, even if we obtain regulatory approvals, the timing or scope of any approvals may prohibit or reduce our ability to commercialize
our product candidates successfully. For example, if the approval process takes too long, we may miss market opportunities and give other
companies the ability to develop competing products or establish market dominance. Any regulatory approval we ultimately obtain may be
limited or subject to restrictions or post-approval commitments that render our product candidates not commercially viable. For example,
regulatory authorities may approve any of our product candidates for fewer or more limited indications than we request, may not approve
the price we intend to charge for any of our product candidates, may grant approval contingent on the performance of costly post-marketing
clinical trials, or may approve any of our product candidates with a label that does not include the labeling claims necessary or desirable
for the successful commercialization of that indication. Further, the FDA or comparable foreign regulatory authorities may place conditions
on approvals or require risk management plans or a Risk Evaluation and Mitigation Strategy, or REMS, to assure the safe use of the drug.
Moreover, product approvals may be withdrawn for non-compliance with regulatory standards or if problems occur following the initial
marketing of the product. Any of the foregoing scenarios could materially harm the commercial success of our product candidates.
Even
if we obtain marketing approval for any of our product candidates, we will be subject to ongoing obligations and continued regulatory
review, which may result in significant additional expense. Additionally, our product candidates could be subject to labeling and other
restrictions and withdrawal from the market and we may be subject to penalties if we fail to comply with regulatory requirements or if
we experience unanticipated problems with our product candidates.
Even
if we obtain regulatory approval for any of our product candidates for an indication, the FDA or foreign equivalent may still impose
significant restrictions on their indicated uses or marketing or the conditions of approval, or impose ongoing requirements for potentially
costly and time-consuming post-approval studies, including Phase 4 clinical trials, and post-market surveillance to monitor safety and
efficacy. Our product candidates will also be subject to ongoing regulatory requirements governing the manufacturing, labeling, packaging,
storage, distribution, safety surveillance, advertising, promotion, recordkeeping and reporting of adverse events and other post-market
information. These requirements include registration with the FDA, as well as continued compliance with current Good Clinical Practices
regulations, or cGCPs, for any clinical trials that we conduct post-approval. In addition, manufacturers of drug products and their facilities
are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with current cGMPs,
requirements relating to quality control, quality assurance and corresponding maintenance of records and documents.
The
FDA has the authority to require a REMS as part of an BLA or after approval, which may impose further requirements or restrictions on
the distribution or use of an approved drug, such as limiting prescribing to certain physicians or medical centers that have undergone
specialized training, limiting treatment to patients who meet certain safe-use criteria or requiring patient testing, monitoring and/or
enrollment in a registry.
With
respect to sales and marketing activities related to our product candidates, advertising and promotional materials must comply with FDA
rules in addition to other applicable federal, state and local laws in the United States and similar legal requirements in other countries.
In the United States, the distribution of product samples to physicians must comply with the requirements of the U.S. Prescription Drug
Marketing Act. Application holders must obtain FDA approval for product and manufacturing changes, depending on the nature of the change.
We may also be subject, directly or indirectly through our customers and partners, to various fraud and abuse laws, including, without
limitation, the U.S. Anti-Kickback Statute, U.S. False Claims Act, and similar state laws, which impact, among other things, our proposed
sales, marketing, and scientific/educational grant programs. If we participate in the U.S. Medicaid Drug Rebate Program, the Federal
Supply Schedule of the U.S. Department of Veterans Affairs, or other government drug programs, we will be subject to complex laws and
regulations regarding reporting and payment obligations. All of these activities are also potentially subject to U.S. federal and state
consumer protection and unfair competition laws. Similar requirements exist in many of these areas in other countries.
In
addition, if any of our product candidates are approved for a particular indication, our product labeling, advertising and promotion
would be subject to regulatory requirements and continuing regulatory review. The FDA strictly regulates the promotional claims that
may be made about prescription products. In particular, a product may not be promoted for uses that are not approved by the FDA as reflected
in the product’s approved labeling. If we receive marketing approval for our product candidates, physicians may nevertheless legally
prescribe our products to their patients in a manner that is inconsistent with the approved label. If we are found to have promoted such
off-label uses, we may become subject to significant liability and government fines. The FDA and other agencies actively enforce the
laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses
may be subject to significant sanctions. The federal government has levied large civil and criminal fines against companies for alleged
improper promotion and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies
enter into consent decrees of permanent injunctions under which specified promotional conduct is changed or curtailed.
If
we or a regulatory agency discover previously unknown problems with a product candidate, such as adverse events of unanticipated severity
or frequency, problems with the facility where the product is manufactured, or we or our manufacturers fail to comply with applicable
regulatory requirements, we may be subject to the following administrative or judicial sanctions:
| |
● |
restrictions
on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary or mandatory product recalls; |
| |
● |
issuance
of warning letters or untitled letters; |
| |
● |
clinical
holds; |
| |
● |
injunctions
or the imposition of civil or criminal penalties or monetary fines; |
| |
● |
suspension
or withdrawal of regulatory approval; |
| |
● |
suspension
of any ongoing clinical trials; |
| |
● |
refusal
to approve pending applications or supplements to approved applications filed by us, or suspension or revocation of product license
approvals; |
| |
● |
suspension
or imposition of restrictions on operations, including costly new manufacturing requirements; or |
| |
● |
product
seizure or detention or refusal to permit the import or export of product. |
The
occurrence of any event or penalty described above may inhibit our ability to commercialize our product candidates and generate revenue.
Adverse regulatory action, whether pre- or post-approval, can also potentially lead to product liability claims and increase our product
liability exposure.
We
also cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative
action, either in the United States or abroad, and compliance with such regulation may be expensive and consume substantial financial
and management resources. If we or any future marketing collaborators or contract manufacturers are slow or unable to adapt to changes
in existing requirements or the adoption of new requirements or policies or are not able to maintain regulatory compliance, it could
delay or prevent the promotion, marketing or sale of our products, which would adversely affect our business and results of operations.
Obtaining
and maintaining regulatory approval of our product candidates in one jurisdiction does not mean that we will be successful in obtaining
regulatory approval of our product candidates in other jurisdictions.
Obtaining
and maintaining regulatory approval of our product candidates in one jurisdiction does not guarantee that we will be able to obtain or
maintain regulatory approval in any other jurisdiction, but a failure or delay in obtaining regulatory approval in one jurisdiction may
have a negative effect on the regulatory approval process in others. For example, even if the FDA grants marketing approval of a product
candidate, comparable regulatory authorities in foreign jurisdictions must also approve the manufacturing, marketing and promotion of
the product candidate in those countries. Approval procedures vary among jurisdictions and can involve requirements and administrative
review periods different from those in the United States, including additional preclinical studies or clinical trials, as clinical studies
conducted in one jurisdiction may not be accepted by regulatory authorities in other jurisdictions. In many jurisdictions outside the
United States, a product candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some
cases, the price that we intend to charge for our products is also subject to approval.
Obtaining
foreign regulatory approvals and compliance with foreign regulatory requirements could result in significant delays, difficulties and
costs for us and could delay or prevent the introduction of our product candidates in certain countries. If we fail to comply with the
regulatory requirements in international markets and/ or to receive applicable marketing approvals, our target market will be reduced
and our ability to realize the full market potential of our product candidates will be harmed.
Even
though we may apply for orphan drug designation for a product candidate, we may not be able to obtain orphan drug marketing exclusivity.
We
believe that in some cases our products candidates may qualify for the FDA’s orphan drug status. There is no guarantee that the
FDA will grant any future application for orphan drug designation for any of our product candidates, which would make us ineligible for
the additional exclusivity and other benefits of orphan drug designation.
Under
the Orphan Drug Act, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition, which is generally
a disease or condition that affects fewer than 200,000 individuals in the United States and for which there is no reasonable expectation
that the cost of developing and making a drug available in the United States for this type of disease or condition will be recovered
from sales of the product. Orphan drug designation must be requested before submitting an BLA. After the FDA grants orphan drug designation,
the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan product designation does
not convey any advantage in or shorten the duration of regulatory review and approval process. In addition to the potential period of
exclusivity, orphan designation makes a company eligible for grant funding of up to $650,000 per year for four years to defray costs
of clinical trial expenses, tax credits for clinical research expenses and potential exemption from the FDA application user fee.
If
a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such
designation, the product is entitled to orphan drug exclusivity, which means the FDA may not approve any other applications to market
the same drug for the same indication for seven years, except in limited circumstances, such as (i) the drug’s orphan designation
is revoked; (ii) its marketing approval is withdrawn; (iii) the orphan exclusivity holder consents to the approval of another applicant’s
product; (iv) the orphan exclusivity holder is unable to assure the availability of a sufficient quantity of drug; or (v) a showing of
clinical superiority to the product with orphan exclusivity by a competitor product. If a drug designated as an orphan product receives
marketing approval for an indication broader than what is designated, it may not be entitled to orphan drug exclusivity. There can be
no assurance that we will receive orphan drug designation for any of our product candidates in the indications for which we think they
might qualify, if we elect to seek such applications.
Current
and future legislation may increase the difficulty and cost for us to obtain marketing approval of and commercialize our product candidates
and affect the prices we may obtain.
In
the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes
regarding the healthcare system that could prevent or delay marketing approval for our product candidates, restrict or regulate post-approval
activities and affect our ability to profitably sell our product candidates. Legislative and regulatory proposals have been made to expand
post-approval requirements and restrict sales and promotional activities for pharmaceutical products. We do not know whether additional
legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact
of such changes on the marketing approvals of our product candidates, if any, may be. In addition, increased scrutiny by the U.S. Congress
of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product
labeling and post-marketing testing and other requirements.
In
the United States, the Medicare Modernization Act, or MMA, changed the way Medicare covers and pays for pharmaceutical products. The
legislation expanded Medicare coverage for drug purchases by the elderly and introduced a new reimbursement methodology based on average
sales prices for drugs. In addition, this legislation authorized Medicare Part D prescription drug plans to use formularies where they
can limit the number of drugs that will be covered in any therapeutic class. As a result of this legislation and the expansion of federal
coverage of drug products, we expect that there will be additional pressure to contain and reduce costs. These cost reduction initiatives
and other provisions of this legislation could decrease the coverage and price that we receive for our product candidates and could seriously
harm our business. While the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage
policy and payment limitations in setting their own reimbursement rates, and any reduction in reimbursement that results from the MMA
may result in a similar reduction in payments from private payors.
The
Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act of 2010 or,
collectively, the ACA, is a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare
spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries,
impose new taxes and fees on the health industry and impose additional health policy reforms. The ACA revised the definition of “average
manufacturer price” for reporting purposes, which could increase the amount of Medicaid drug rebates to states. The law also imposed
a significant annual fee on companies that manufacture or import branded prescription drug products. Further, on August 16, 2022, President
Biden signed the Inflation Reduction Act of 2022, or IRA, into law which, among other things, extends enhanced subsidies for individuals
purchasing health insurance coverage in ACA marketplaces through plan year 2025. The IRA also eliminates the “donut hole”
under the Medicare Part D program beginning in 2025 by significantly lowering the beneficiary maximum out-of-pocket cost and creating
a new manufacturer discount program. It is unclear how such challenges, and the healthcare reform measures of the Biden administration
will impact the ACA and our business.
In
addition, other legislative changes have been proposed and adopted in the United States since the ACA was enacted. In 2011, the U.S.
Congress enacted the Budget Control Act of 2011, or the Budget Control Act, which included provisions intended to reduce the federal
deficit. The Budget Control Act resulted in the imposition of 2% reductions in Medicare payments to providers beginning in 2013 and,
due to subsequent legislative amendments to the statute, will remain in effect through 2027 absent additional congressional action. However,
pursuant to the CARES Act, and subsequent legislation, these reductions are suspended from May 1, 2020 through March 31, 2022 due to
the COVID-19 pandemic. In January 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, reduced
Medicare payments to several providers, and increased the statute of limitations period for the government to recover overpayments to
providers from three to five years. These new laws may result in additional reductions in Medicare and other healthcare funding, which
could have a material adverse effect on customers for our drugs, if approved, and accordingly, our financial operations. If government
spending is further reduced, anticipated budgetary shortfalls may also impact the ability of relevant agencies, such as the FDA, to continue
to function at current levels, which may impact the ability of relevant agencies to timely review and approve research and development,
manufacturing and marketing activities, which may delay our ability to develop, market and sell any product candidates we may develop.
In addition, any significant spending reductions affecting Medicare, Medicaid or other publicly funded or subsidized health programs
that may be implemented, or any significant taxes or fees that may be imposed on us, as part of any broader deficit reduction effort
or legislative replacement to the Budget Control Act, could have an adverse impact on our anticipated product revenues.
Moreover,
recently there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their commercial products.
There have been several recent U.S. Congressional inquiries and proposed and enacted federal and state legislation designed to, among
other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, reduce
the cost of drugs under Medicare, and reform government program reimbursement methodologies for drugs. On September 24, 2020, the FDA
released a final rule providing guidance for states to build and submit importation plans for drugs from Canada. Further, on November
20, 2020, the U.S. Department of Health and Human Services, or HHS, finalized a regulation removing safe harbor protection for price
reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers, unless
the price reduction is required by law. The rule also creates a new safe harbor for price reductions reflected at the point-of-sale,
as well as a safe harbor for certain fixed fee arrangements between pharmacy benefit managers and manufacturers. On August 16, 2022,
Congress enacted the Inflation Reduction Act of 2022 which contains several provisions relating to prescription drug costs, including
requirements for federal government price negotiations, rebate requirements, and caps on out-of-pocket spending for Medicare Part D enrollees.
At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical
and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access
and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and
bulk purchasing.
On
November 20, 2020, the HHS Office of Inspector General finalized further modifications to the federal Anti-Kickback Statute. Under the
final rules, the HHS Office of Inspector General added safe harbor protections under the Anti-Kickback Statute for certain coordinated
care and value-based arrangements among clinicians, providers, and others, yet removed safe harbor protection for price reductions from
pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers, unless the price reduction
is required by law. The rule also creates a new safe harbor for price reductions reflected at the point-of-sale, as well as a safe harbor
for certain fixed fee arrangements between pharmacy benefit managers and manufacturers. This rule (with exceptions) became effective
January 19, 2021. We continue to evaluate what effect, if any, these rules will have on our business. CMS issued a final rule, effective
on July 9, 2019, that requires direct-to-consumer advertisements of prescription drugs and biological products, for which payment is
available through or under Medicare or Medicaid, to include in the advertisement the Wholesale Acquisition Cost, or list price, of that
drug or biological product if it is equal to or greater than $35 for a monthly supply or usual course of treatment. Prescription drugs
and biological products that are in violation of these requirements will be included on a public list. Any adopted health reform measure
could reduce the ultimate demand for our products, if approved, or put pressure on our product pricing. Individual states in the United
States have also become increasingly active in passing legislation and implementing regulations designed to control pharmaceutical product
pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure
and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. In addition,
regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products
and which suppliers will be included in their prescription drug and other healthcare programs. We expect that additional state and federal
healthcare reform measures will be adopted in the future.
The
delivery of healthcare in the European Union, or EU, including the establishment and operation of health services and the pricing and
reimbursement of medicines, is almost exclusively a matter for national, rather than EU, law and policy. National governments and health
service providers have different priorities and approaches to the delivery of health care and the pricing and reimbursement of products
in that context. In general, however, the healthcare budgetary constraints in most EU member states have resulted in restrictions on
the pricing and reimbursement of medicines by relevant health service providers. Coupled with ever-increasing EU and national regulatory
burdens on those wishing to develop and market products, this could prevent or delay marketing approval of our product candidates, restrict
or regulate post-approval activities and affect our ability to commercialize any products for which we obtain marketing approval. Both
in the United States and in the EU, legislative and regulatory proposals have been made to expand post-approval requirements and restrict
sales and promotional activities for pharmaceutical products. We do not know whether additional legislative changes will be enacted,
or whether the regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals
of our product candidates, if any, may be.
Third-party
coverage and reimbursement and health care cost containment initiatives and treatment guidelines may constrain our future revenues.
Our
ability to successfully market our product candidates will depend in part on the level of reimbursement that government health administration
authorities, private health coverage insurers and other organizations provide for the cost of our product candidates and related treatments.
Countries in which any of our product candidates are sold through reimbursement schemes under national health insurance programs frequently
require that manufacturers and sellers of pharmaceutical products obtain governmental approval of initial prices and any subsequent price
increases. In certain countries, including the United States, government-funded and private medical care plans can exert significant
indirect pressure on prices. We may not be able to sell our product candidates profitably if adequate prices are not approved or coverage
and reimbursement is unavailable or limited in scope. Increasingly, third-party payors attempt to contain health care costs in ways that
are likely to impact our development of products including:
| |
● |
failing
to approve or challenging the prices charged for health care products; |
| |
● |
introducing
reimportation schemes from lower priced jurisdictions; |
| |
● |
limiting
both coverage and the amount of reimbursement for new therapeutic products; |
| |
● |
denying
or limiting coverage for products that are approved by the regulatory agencies but are considered to be experimental or investigational
by third-party payors; and |
| |
● |
refusing
to provide coverage when an approved product is used in a way that has not received regulatory marketing approval. |
Our
relationships with customers and payors are subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations,
which, if violated, could expose us to criminal sanctions, civil penalties, exclusion from government healthcare programs, contractual
damages, reputational harm and diminished profits and future earnings.
Healthcare
providers, physicians and others will play a primary role in the recommendation and prescription of any products for which we obtain
marketing approval. Our future arrangements with third-party payors and customers may expose us to broadly applicable fraud and abuse
and other healthcare laws and regulations, primarily in the United States, that may constrain the business or financial arrangements
and relationships through which we market, sell and distribute our products for which we obtain marketing approval. Restrictions under
applicable healthcare laws and regulations, include the following:
| |
● |
the
federal Anti-Kickback Statute prohibits, among other things, the knowing and willful offer, payment, solicitation or receipt of any
form of remuneration in return for, or to induce, (i) the referral of a person, (ii) the furnishing or arranging for the furnishing
of items or services reimbursable under the Medicare, Medicaid or other governmental programs, or (iii) the purchase, lease or order
or arranging or recommending purchasing, leasing or ordering of any item or service reimbursable under the Medicare, Medicaid or
other governmental programs. A person or entity does not need to have actual knowledge of the statute or specific intent to violate
it in order to have committed a violation. In addition, the government may assert that a claim including items or services resulting
from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act; |
| |
● |
the
federal False Claims Act imposes civil penalties, and provides for civil whistleblower or qui tam actions, against individuals or
entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent
or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government; |
| |
● |
HIPAA
imposes criminal and civil liability for executing a scheme to defraud any health care benefit program or making false statements
relating to health care matters. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge
of the statute or specific intent to violate it in order to have committed a violation; |
| |
● |
the
federal false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making
any materially false statement in connection with the delivery of or payment for health care benefits, items or services; |
| |
● |
the
Physician Payments Sunshine Act, created under the ACA, and its implementing regulations, which requires specified manufacturers
of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s
Health Insurance Program, with specific exceptions, to report annually to CMS information related to payments or other “transfers
of value” made to physicians. All such reported information is publicly available; |
| |
● |
analogous
state and non-U.S. laws and regulations, such as certain state anti-kickback and false claims laws which may apply to items or services
reimbursed by any third-party payor, including commercial insurers; state laws that require pharmaceutical companies to comply with
the industry’s voluntary compliance guidelines and the applicable compliance guidance promulgated by the federal government
or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws that require
drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers
or marketing expenditures; and state laws governing the privacy and security of health information in certain circumstances, many
of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts; and |
| |
● |
regulation
by CMS and enforcement by the HHS Office of Inspector General or the U.S. Department of Justice. |
Because
of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our
future business activities could be subject to challenge under one or more of such laws.
Efforts
to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve
substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current
or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations
are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant
civil, criminal and administrative penalties, damages, fines, exclusion from U.S. government funded healthcare programs, such as Medicare
and Medicaid, and the curtailment or restructuring of our operations. If any of the physicians or other providers or entities with whom
we expect to do business with are found to be not in compliance with applicable laws, they may be subject to criminal, civil or administrative
sanctions, including exclusions from government funded healthcare programs.
Risks
Relating to Our Intellectual Property Rights
It
is difficult and costly to protect our intellectual property rights, and we cannot ensure the protection of these rights.
Our
commercial success will depend, in part, on our ability to prosecute and defend, if necessary, our patent rights against third-party
challenges and successfully enforcing these patent rights against third party competitors. The patent positions of biotech companies
can be highly uncertain and involve complex legal, scientific and factual questions for which important legal principles remain unresolved.
Changes in either the patent laws or in interpretations of patent laws may diminish the value of our intellectual property. Accordingly,
we cannot predict the breadth of claims that may be allowable or enforceable in any patent applications filed by us or our licensors
of patent rights. The patents and patent applications held by or licensed to us relating to our microbial platform and related technologies
may be challenged, invalidated or circumvented by third parties and might not protect us against competitors with similar products or
technologies.
The
degree of future protection afforded by the patent rights held by or licensed to us is uncertain, because legal means afford only limited
protection and may not adequately protect our rights, permit us to gain or keep our competitive advantage, or provide us with any competitive
advantage at all. We cannot be certain that any patent application owned by a third party will not have priority over patent applications
filed by us or in which we hold license rights or that we will not be involved in interference, opposition or invalidity proceedings
before United States or foreign patent offices.
Additionally,
if we were to initiate legal proceedings against a third party to enforce a patent covering any of our product candidates, the defendant
could counterclaim that such patent is invalid and/or unenforceable. In patent litigation in the United States, defendant counterclaims
alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge include alleged failures to meet any of
several statutory requirements, including lack of novelty, obviousness or non-enablement. Grounds for unenforceability assertions include
allegations that someone connected with prosecution of the patent withheld relevant information from the United States Patent and Trademark
Office, or the USPTO, or made a misleading statement, during prosecution. Third parties may also raise similar claims before administrative
bodies in the United States or abroad, even outside the context of litigation. Such mechanisms include re-examination, post grant review
and equivalent proceedings in foreign jurisdictions, e.g., opposition proceedings. Such proceedings could result in revocation or amendment
of patents held by or licensed to us in such a way that they no longer cover our product candidates or competitive products. The outcome
following legal assertions of invalidity and unenforceability is unpredictable. With respect to validity, for example, we cannot be certain
that there is no invalidating prior art, of which, we, any licensor of our patent rights or the patent examiner were unaware during prosecution.
If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all,
of the patent protection on any of our product candidates. Such a loss of patent protection would have a material adverse impact on our
business.
We
rely on know-how and trade secrets to protect technology, especially in cases where we believe patent protection is not appropriate or
obtainable. However, know-how and trade secrets are difficult to protect. While we require employees, academic collaborators, consultants
and other contractors to enter into confidentiality agreements, we may not be able to adequately protect our trade secrets or other proprietary
or licensed information. Typically, research collaborators and scientific advisors have rights to publish data and information in which
we may have rights. Enforcing a claim that a third party illegally obtained and is using any of our trade secrets is expensive and time
consuming, and the outcome is unpredictable. In addition, courts are sometimes less willing to protect trade secrets than patents. Moreover,
our competitors may independently develop equivalent knowledge, methods and know-how.
If
we fail to obtain or maintain patent protection or trade secret protection for our product candidates or our technologies, third parties
could use our proprietary information, which could impair our ability to compete in the market and adversely affect our ability to generate
revenues and attain profitability.
Our
product candidates may infringe the intellectual property rights of others, which could increase our costs and delay or prevent our development
and commercialization efforts.
Our
success depends in part on avoiding infringement of the proprietary technologies of others. The pharmaceutical industry has been characterized
by frequent litigation regarding patent and other intellectual property rights. Identification of third-party patent rights that may
be relevant to our proprietary technology is difficult because patent searching is imperfect due to differences in terminology among
patents, incomplete databases and the difficulty in assessing the meaning of patent claims. Additionally, because patent applications
are maintained in secrecy until the application is published, we may be unaware of third-party patents that may be infringed by commercialization
of any of our product candidates or any future product candidate. There may be certain issued patents and patent applications claiming
subject matter that we may be required to license in order to research, develop or commercialize any of our product candidates, and we
do not know if such patents and patent applications would be available to license on commercially reasonable terms, or at all. Any claims
of patent infringement asserted by third parties would be time-consuming and may:
| |
● |
result
in costly litigation; |
| |
● |
divert
the time and attention of our technical personnel and management; |
| |
● |
prevent
us from commercializing a product until the asserted patent expires or is held finally invalid or not infringed in a court of law; |
| |
● |
require
us to cease or modify our use of the technology and/or develop non-infringing technology; or |
| |
● |
require
us to enter into royalty or licensing agreements. |
Third
parties may hold proprietary rights that could prevent any of our product candidates from being marketed. Any patent-related legal action
against us claiming damages and seeking to enjoin commercial activities relating to any of our product candidates or our processes could
subject us to potential liability for damages and require us to obtain a license to continue to manufacture or market any of our product
candidates or any future product candidates. We cannot predict whether we would prevail in any such actions or that any license required
under any of these patents would be made available on commercially acceptable terms, if at all. In addition, we cannot be sure that we
could redesign our product candidates or any future product candidates or processes to avoid infringement, if necessary. Accordingly,
an adverse determination in a judicial or administrative proceeding, or the failure to obtain necessary licenses, could prevent us from
developing and commercializing any of our product candidates or a future product candidate, which could harm our business, financial
condition and operating results.
We
expect that there are other companies, including major biopharmaceutical companies, working in the areas competitive to our proposed
product candidates which either has resulted, or may result, in the filing of patent applications that may be deemed related to our activities.
If we were to challenge the validity of these or any issued United States patent in court, we would need to overcome a statutory presumption
of validity that attaches to every issued United States patent. This means that, in order to prevail, we would have to present clear
and convincing evidence as to the invalidity of the patent’s claims. If we were to challenge the validity of these or any issued
United States patent in an administrative trial before the Patent Trial and Appeal Board in the USPTO, we would have to prove that the
claims are unpatentable by a preponderance of the evidence. There is no assurance that a jury and/or court would find in our favor on
questions of infringement, validity or enforceability. Even if we are successful, litigation could result in substantial costs and be
a distraction to management.
We
may be subject to claims that we have wrongfully hired an employee from a competitor or that we or our employees have wrongfully used
or disclosed alleged confidential information or trade secrets of their former employers.
As
is commonplace in our industry, we will employ individuals who were previously employed at other biopharmaceutical companies, including
our competitors or potential competitors. Although no claims against us are currently pending, we may be subject in the future to claims
that our employees or prospective employees are subject to a continuing obligation to their former employers (such as non-competition
or non-solicitation obligations) or claims that our employees or we have inadvertently or otherwise used or disclosed trade secrets or
other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful
in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Risks
Related to this Offering and Owning Our Common Stock and/or Pre-Funded Warrants
The
market price of our shares may be subject to fluctuation and volatility. You could lose all or part of your investment.
The
market price of our common stock is subject to wide fluctuations in response to various factors, some of which are beyond our control.
Since shares of our common stock were sold in our initial public offering, or IPO, in June 2023 at a price of $5.00 per share, the reported
high and low sales prices of our common stock have ranged from $5.18 to $0.92 through January 12, 2024. The market price
of our shares on the NYSE American may fluctuate as a result of a number of factors, some of which are beyond our control, including,
but not limited to:
| |
● |
actual
or anticipated variations in our and our competitors’ results of operations and financial condition; |
| |
● |
changes
in earnings estimates or recommendations by securities analysts, if our shares are covered by analysts; |
| |
● |
market
acceptance of our product candidates; |
| |
● |
development
of technological innovations or new competitive products by others; |
| |
● |
announcements
of technological innovations or new products by us; |
| |
● |
publication
of the results of preclinical or clinical trials for our product candidates; |
| |
● |
failure
by us to achieve a publicly announced milestone; |
| |
● |
delays
between our expenditures to develop and market new or enhanced products and the generation of sales from those products; |
| |
● |
developments
concerning intellectual property rights, including our involvement in litigation brought by or against us; |
| |
● |
regulatory
developments and the decisions of regulatory authorities as to the approval or rejection of new or modified products; |
| |
● |
changes
in the amounts that we spend to develop, acquire or license new products, technologies or businesses; |
| |
● |
changes
in our expenditures to promote our product candidates; |
| |
● |
our
sale or proposed sale, or the sale by our significant stockholders, of our shares or other securities in the future; |
| |
● |
changes
in key personnel; |
| |
● |
success
or failure of our research and development projects or those of our competitors; |
| |
● |
the
trading volume of our shares; and |
| |
● |
general
economic and market conditions and other factors, including factors unrelated to our operating performance. |
These
factors and any corresponding price fluctuations may materially and adversely affect the market price of our shares and result in substantial
losses being incurred by our investors. In the past, following periods of market volatility, public company stockholders have often instituted
securities class action litigation. If we were involved in securities litigation, it could impose a substantial cost upon us and divert
the resources and attention of our management from our business.
There
is no public market for the Pre-Funded Warrants being offered in this offering.
There
is no established public trading market for the Pre-Funded Warrants being offered in this offering, and we do not expect a market to
develop. In addition, we do not intend to apply to list the Pre-Funded Warrants on any securities exchange or nationally recognized trading
system, including NYSE American. Without an active market, the liquidity of the Pre-Funded Warrants will be limited.
We
will not receive any meaningful amount of additional funds upon the exercise of the Pre-Funded Warrants.
Each
Pre-Funded Warrant will be exercisable until it is fully exercised and by means of payment of the nominal cash purchase price upon exercise.
Accordingly, we will not receive any meaningful additional funds upon the exercise of the Pre-Funded Warrants.
Holders
of the Pre-Funded Warrants purchased in this offering will have no rights as common stockholders until such holders exercise such warrants
and acquire shares of our common stock.
Until
holders of the Pre-Funded Warrants acquire shares of our common stock upon exercise thereof, holders of such Pre-Funded Warrants will
have no rights with respect to the shares of our common stock underlying such Pre-Funded Warrants. Upon exercise of the Pre-Funded Warrants,
such holders will be entitled to exercise the rights of a common stockholder only as to matters for which the record date occurs after
the exercise date.
Significant
holders or beneficial holders of shares of our common stock may not be permitted to exercise the Pre-Funded Warrants that they hold.
A
holder of the Pre-Funded Warrants will not be entitled to exercise any portion of any Pre-Funded Warrant that, upon giving effect to
such exercise, would cause: (i) the aggregate number of shares of our common stock beneficially owned by such holder (together with its
affiliates) to exceed 4.99% (or, upon election of holder, 9.99%) of the number of shares of our common stock outstanding immediately
after giving effect to the exercise; or (ii) the combined voting power of our securities beneficially owned by such holder (together
with its affiliates) to exceed 4.99% (or, upon election of holder, 9.99%) of the combined voting power of all of our securities outstanding
immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the Pre-Funded
Warrants. As a result, you may not be able to exercise your Pre-Funded Warrants for shares of our common stock at a time when it would
be financially beneficial for you to do so. In such a circumstance, you could seek to sell your Pre-Funded Warrants to realize value,
but you may be unable to do so in the absence of an established trading market and due to applicable transfer restrictions.
Our
failure to meet the continued listing requirements of the NYSE American could result in a delisting of our common stock.
If
we fail to satisfy the continued listing requirements of the NYSE American, such as the corporate governance requirements or the minimum
closing bid price requirement, the NYSE American may take steps to delist our common stock. Such a delisting would likely have a negative
effect on the price of our common stock and would impair your ability to sell or purchase our common stock when you wish to do so. In
the event of a delisting, we can provide no assurance that any action taken by us to restore compliance with listing requirements would
allow our common stock to become listed again, stabilize the market price or improve the liquidity of our common stock, prevent our common
stock from dropping below the NYSE American’s minimum bid price requirement or prevent future non-compliance with the NYSE American’s
listing requirements.
Future
capital raises may dilute your ownership and/or have other adverse effects on our operations.
If
we raise additional capital by issuing equity securities, our existing stockholders’ percentage ownership will be reduced and these
stockholders may experience substantial dilution. If we raise additional funds by issuing debt securities, these debt securities would
have rights senior to those of our common stock and the terms of the debt securities issued could impose significant restrictions on
our operations, including liens on our assets. If we raise additional funds through collaborations and licensing arrangements, we may
be required to relinquish some rights to our intellectual property or candidate products, or to grant licenses on terms that are not
favorable to us.
Shares
eligible for future sale may adversely affect the market for our common stock.
As
of the date of this prospectus, we have 12,097,643 shares of common stock issued and outstanding, all of which are eligible for sale
by means of ordinary brokerage transactions in the open market pursuant to Rule 144, promulgated under the Securities Act, subject to
certain limitations under Rule 144, except for approximately 588,745 shares of common stock held by our officers and directors which
are subject to IPO lock-ups expiring in June 2024. We have granted demand and piggyback registration rights to the former holders of
our convertible preferred stock and convertible promissory notes pursuant to which they may request the registration for resale of up
to 9,542,519 shares of common stock. Any substantial sale of our common stock pursuant to Rule 144 or pursuant to any resale prospectus
(including sales by investors of securities acquired in connection with this offering) may have a material adverse effect on the market
price of our common stock.
We
are an “emerging growth company” under the JOBS Act and we cannot be certain if the reduced disclosure requirements applicable
to emerging growth companies will make our common stock less attractive to investors.
We
are an “emerging growth company,” as defined in the JOBS Act and we may take advantage of certain exemptions from various
reporting requirements that are applicable to other public companies that are not “emerging growth companies” including,
but not limited to:
| |
● |
not
being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act; |
| |
● |
reduced
disclosure obligations regarding executive compensation in our periodic reports and proxy statements; |
| |
● |
exemptions
from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute
payments; and |
| |
● |
extended
transition periods available for complying with new or revised accounting standards. |
We
have chosen to take advantage of all of the benefits available under the JOBS Act, including the exemptions discussed above. We cannot
predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common
stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
We
will remain an “emerging growth company” for up to five years, although we will lose that status sooner if our revenues exceed
$1.235 billion, if we issue more than $1 billion in non-convertible debt in a three-year period, or if the market value of our common
stock that is held by non-affiliates exceeds $700 million as of June 30 in any future year.
Our
status as an “emerging growth company” under the JOBS Act may make it more difficult to raise capital as and when we need
it.
Because
of the exemptions from various reporting requirements provided to us as an “emerging growth company,” we may be less attractive
to investors and it may be difficult for us to raise additional capital as and when we need it. Investors may be unable to compare our
business with other companies in our industry if they believe that our reporting is not as transparent as other companies in our industry.
If we are unable to raise additional capital as and when we need it, our financial condition and results of operations may be materially
and adversely affected.
We
have not paid dividends on our common stock in the past and have no immediate plans to pay such dividends.
We
plan to reinvest all of our earnings, to the extent we have earnings, to cover operating costs and otherwise become and remain competitive.
We do not plan to pay any cash dividends with respect to our common stock in the foreseeable future. We cannot assure you that we would,
at any time, generate sufficient surplus cash that would be available for distribution to the holders of our common stock as a dividend.
Therefore, you should not expect to receive cash dividends on the common stock we are offering.
If
equity research analysts do not publish research or reports about our business or if they issue unfavorable commentary or downgrade our
shares, the price of our shares could decline.
The
trading market for our shares will rely in part on the research and reports that equity research analysts publish about us and our business,
if at all. We do not have control over these analysts and we do not have commitments from them to write research reports about us. The
price of our shares could decline if no research reports are published about us or our business, or if one or more equity research analysts
downgrades our shares or if those analysts issue other unfavorable commentary or cease publishing reports about us or our business.
You
will experience immediate dilution in the book value per share of the common stock or Pre-Funded Warrant you purchase.
Because
the price per share of our common stock and Pre-Funded Warrants being offered is substantially higher than the book value per
share of our common stock, you will experience substantial dilution in the net tangible book value of the common stock and/or Pre-Funded
Warrants you purchase in this offering. Based on the public offering price of $ per share, if you purchase shares of common stock
and/or Pre-Funded Warrants in this offering, you will experience immediate and substantial dilution of $1.36 per share
in the net tangible book value of the common stock at September 30, 2023.
We
may be at an increased risk of securities class action litigation.
Historically,
securities class action litigation has often been brought against a company following a decline in the market price of its securities.
This risk is especially relevant for us because biotechnology and pharmaceutical companies have experienced significant stock price volatility
in recent years. If we were to be sued, it could result in substantial costs and a diversion of management’s attention and resources,
which could harm our business. We maintain director and officer insurance that we regard as reasonably adequate to protect us from potential
claims; however, we are responsible for meeting certain deductibles under the policies and, in any event, we cannot assure you that the
insurance coverage will adequately protect us from claims made. Further, the costs of insurance may increase and the availability of
coverage may decrease. As a result, we may not be able to maintain our current levels of insurance at a reasonable cost, or at all.
We
have broad discretion in how we use the proceeds of this offering and may not use these proceeds effectively, which could affect our
results of operations and cause our stock price to decline.
We
may invest or spend these proceeds in ways with which you do not agree and in ways that may not yield a return on your investment. Our
management will have considerable discretion in the application of the net proceeds of this offering, including for any purpose described
in the section of this prospectus entitled “Use of Proceeds”. However, our needs may change as our business and industry
evolve and, as a result, the proceeds we receive from this offering may be used in a manner substantially different from our current
expectations. We may use the net proceeds for purposes that do not yield a significant return or any return at all for our stockholders.
In addition, pending their use, we may invest the net proceeds from this offering in a manner that does not produce income or that loses
value. The failure by our management to apply these funds effectively could result in financial losses that could harm our business,
cause the price of our common stock to decline and delay the development of our product candidates. You will not have the opportunity
as part of your investment decision to assess whether the net proceeds are being used appropriately and, as a result, you will be relying
on our management’s judgment.
Our
charter documents and Delaware law may inhibit a takeover that stockholders consider favorable.
Provisions
of our amended and restated certificate of incorporation and amended and restated bylaws and applicable provisions of Delaware law may
delay or discourage transactions involving an actual or potential change in control or change in our management, including transactions
in which stockholders might otherwise receive a premium for their shares, or transactions that our stockholders might otherwise deem
to be in their best interests. The provisions in our amended and restated certificate of incorporation and amended and restated bylaws:
| |
● |
limit
who may call stockholder meetings; |
| |
● |
restrict
our stockholders from bringing matters before our annual meeting of stockholders or from making nominations for directors at our
annual meeting of stockholders if the proper procedures are not followed; |
| |
● |
do
not provide for stockholder action by written consent; |
| |
● |
do
not provide for cumulative voting rights; and |
| |
● |
provide
that all board vacancies may be filled by the affirmative vote of a majority of directors then in office, even if less than a quorum. |
Section
203 of the DGCL may limit our ability to engage in any business combination with a person who beneficially owns 15% or more of our outstanding
voting stock unless certain conditions are satisfied. This restriction lasts for a period of three years following the share acquisition.
These provisions may have the effect of entrenching our management team and may deprive you of the opportunity to sell your shares to
potential acquirers at a premium over prevailing prices. This potential inability to obtain a control premium could reduce the price
of our common stock.
General
Risk Factors
Changes
in accounting standards and subjective assumptions, estimates and judgments by management related to complex accounting matters may materially
impact reporting of our financial condition and results of operations.
Accounting
principles generally accepted in the United States and related accounting pronouncements, implementation guidelines, and interpretations
we apply to a wide range of matters that are relevant to our business, such as accounting for long-lived asset impairment and share-based
compensation, are complex and involve subjective assumptions, estimates and judgments by our management. Changes in these rules or their
interpretation or changes in underlying assumptions, estimates or judgments by our management could significantly change or add significant
volatility to our reported or expected financial performance.
A
potential failure to maintain effective internal control over financial reporting in accordance with Section 404 of the Sarbanes-Oxley
Act could have a material adverse effect on our business, financial condition, and results of operations.
Our
management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over
financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation
of financial statements in accordance with U.S. generally accepted accounting principles, or GAAP. Under standards established by the
Public Company Accounting Oversight Board, or PCAOB, a deficiency in internal control over financial reporting exists when the design
or operation of a control does not allow management or personnel, in the normal course of performing their assigned functions, to prevent
or detect misstatements on a timely basis. The PCAOB defines a material weakness as a deficiency, or combination of deficiencies, in
internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of annual or interim
financial statements will not be prevented, or detected and corrected, on a timely basis.
We
are required, pursuant to Section 404 of the Sarbanes-Oxley Act, to furnish a report by management on, among other things, the effectiveness
of our internal control over financial reporting for our second annual report on Form 10-K filed with the SEC and in each year thereafter.
Our auditors will also need to attest to the effectiveness of our internal control over financial reporting at such time as we are an
accelerated filed or large accelerated filer and no longer an emerging growth company or smaller reporting company. If we are unable
to assert that our internal control over financial reporting is effective, or when required in the future, if our independent registered
public accounting firm is unable to express an unqualified opinion as to the effectiveness of our internal control over financial reporting,
investors may lose confidence in the accuracy and completeness of our financial reports, the market price of our common stock could be
adversely affected, and we could become subject to litigation or investigations by the stock exchange on which our common stock are listed,
the SEC or other regulatory authorities, which could require additional financial and management resources and could have a material
adverse effect on our business, financial condition, and results of operations.
The
limited amount of public company experience of our management team could adversely impact our ability to comply with the reporting requirements
of U.S. securities laws, which could have a materially adverse effect on our business.
Our
officers have limited public company experience, which could impair our ability to comply with legal and regulatory requirements such
as those imposed by Sarbanes-Oxley Act. Such responsibilities include complying with federal securities laws and making required disclosures
on a timely basis. Any such deficiencies, weaknesses or lack of compliance could have a materially adverse effect on our ability to comply
with the reporting requirements of the Securities Exchange Act of 1934, or the Exchange Act, which is necessary to maintain our public
company status. If we were to fail to fulfill those obligations, our ability to continue as a U.S. public company would be in jeopardy
in which event you could lose your entire investment in our Company.
We
identified material weaknesses in our internal control over financial reporting, and we may identify additional material weaknesses in
the future that may cause us to fail to meet our reporting obligations or result in material misstatements of our financial statements.
If we fail to remediate any material weaknesses or if we otherwise fail to establish and maintain effective control over financial reporting,
our ability to accurately and timely report our financial results could be adversely affected.
Effective
internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure
controls and procedures, are designed to prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered
in their implementation, could cause us to fail to meet our reporting obligations. In addition, any testing by us conducted in connection
with Section 404 of the Sarbanes-Oxley Act, or the subsequent testing by our independent registered public accounting firm when required,
may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses or that may require
prospective or retrospective changes to our financial statements or identify other areas for further attention or improvement. Inferior
internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect
on the trading price of our shares of common stock. There is also a risk that neither we nor our independent registered public accounting
firm (when applicable in the future) will be able to conclude within the prescribed timeframe that internal controls over financial reporting
is effective as required by Section 404. As a result, investors could lose confidence in our financial and other public reporting, which
would harm our business and the trading price of our common stock.
During
the preparation of our financial statements included in this prospectus, we and our independent registered public accounting firm identified
a material weakness as it relates to a lack of adequate segregation of accounting functions. We are in the process of implementing measures
designed to improve our internal control over financial reporting and remediate this material weakness. We intend to increase staffing
within our accounting infrastructure sufficient to facilitate proper segregation of accounting functions.
We
may identify future material weaknesses in our internal controls over financial reporting or fail to meet the demands that will be placed
upon us as a public company, including the requirements of the Sarbanes-Oxley, and we may be unable to accurately report our financial
results, or report them within the timeframes required by law or stock exchange regulations. We cannot assure that our existing material
weakness will be remediated or that additional material weaknesses will not exist or otherwise be discovered, any of which could adversely
affect our reputation, financial condition and results of operations.
We
have and will continue to incur significant increased costs as a result of being a public company that reports to the SEC and our management
will be required to devote substantial time to meet compliance obligations.
As
a public company reporting to the SEC after this offering, we will continue to incur significant legal, accounting and other expenses
that we did not incur as a private company. We are subject to reporting requirements of the Exchange Act and the reporting and governance
provisions of the Sarbanes-Oxley Act and the Dodd-Frank Wall Street Reform and Protection Act, as well as rules subsequently implemented
by the SEC, that impose significant requirements on public companies, including requiring establishment and maintenance of effective
disclosure and financial controls and changes in corporate governance practices. There are significant corporate governance and reporting
provisions in these laws that will increase our legal and financial compliance costs, make some activities more difficult, time-consuming
or costly and may also place undue strain on our personnel, systems and resources. Our management and other personnel will need to devote
a substantial amount of time to these regulations. In addition, we expect these rules and regulations to make it more difficult and more
expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced policy limits and coverage
or incur substantially higher costs to obtain the same or similar coverage. As a result, it may be more difficult for us to attract and
retain qualified people to serve on our Board our Board committees or as executive officers.
Unfavorable
geopolitical and macroeconomic developments could adversely affect our business, financial condition or results of operations.
Our
business could be adversely affected by conditions in the U.S. and global economies, the United States and global financial markets and
adverse geopolitical and macroeconomic developments, including rising inflation rates, the ongoing COVID-19 pandemic, the Ukrainian/Russian
and Israeli/Palestinian conflicts and related sanctions, bank failures, and economic uncertainties related to these conditions.
For
example, inflation rates, particularly in the United States, have increased recently to levels not seen in years, and increased inflation
may result in increases in our operating costs (including our labor costs), reduced liquidity and limits on our ability to access credit
or otherwise raise capital on acceptable terms, if at all. In response to rising inflation, the U.S. Federal Reserve has raised, and
may again raise, interest rates, which, coupled with reduced government spending and volatility in financial markets, may have the effect
of further increasing economic uncertainty and heightening these risks. The failures of Silvergate Bank, Silicon Valley Bank and Signature
Bank in March 2023 and First Republic Bank in May 2023, and fears of other bank failures, may exacerbate these risks. A weak or declining
economy could also strain our suppliers and manufacturers, possibly resulting in supply disruption. Any of the foregoing could harm our
business.
Additionally,
financial markets around the world experienced volatility following the invasion of Ukraine by Russia in February 2022 and the eruption
of the Israeli/Palestinian conflict in October 2023, including as a result of economic sanctions and export controls against Russia and
countermeasures taken by Russia. The full economic and social impact of these sanctions and countermeasures, in addition to the ongoing
military conflicts in Ukraine and Gaza, which could conceivably expand, remains uncertain; however, both the conflicts and related sanctions
have resulted and could continue to result in disruptions to trade, commerce, pricing stability, credit availability, and/or supply chain
continuity, in both Europe and globally, and has introduced significant uncertainty into global markets. While we do not currently operate
in Russia, Ukraine or the Middle East, as the adverse effects of these conflicts continue to develop our business and results of operations
may be adversely affected.
CAUTIONARY
NOTE REGARDING FORWARD-LOOKING STATEMENTS
This
prospectus, including the sections entitled “Prospectus Summary,” “Risk Factors,” “Use
of Proceeds,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations,”
and “Business,” contains forward-looking statements. The words “believe,” “may,” “will,”
“potentially,” “estimate,” “continue,” “anticipate,” “intend,” “could,”
“would,” “project,” “plan,” “expect” and similar expressions that convey uncertainty
of future events or outcomes are intended to identify forward-looking statements. These forward-looking statements include, but are not
limited to, statements concerning the following:
| |
● |
our
future financial and operating results; |
| |
● |
our
intentions, expectations and beliefs regarding anticipated growth, market penetration and trends in our business; |
| |
● |
the
timing and success of our plan of commercialization; |
| |
● |
our
ability to successfully develop and clinically test our product candidates; |
| |
● |
our
ability to obtain FDA approval for any of our product candidates; |
| |
● |
our
ability to comply with all U.S. and foreign regulations concerning the development, manufacture and sale of our product candidates; |
| |
● |
our
reliance on third parties to manufacture our product candidates; |
| |
● |
the
adequacy of the net proceeds of this offering; |
| |
● |
the
effects of market conditions on our stock price and operating results; |
| |
● |
our
ability to maintain, protect and enhance our intellectual property; |
| |
● |
the
effects of increased competition in our market and our ability to compete effectively; |
| |
● |
our
plans to use the proceeds from this offering; |
| |
● |
costs
associated with initiating and defending intellectual property infringement and other claims; |
| |
● |
the
attraction and retention of qualified employees and key personnel; |
| |
● |
future
acquisitions of or investments in complementary companies or technologies; and |
| |
● |
our
ability to comply with evolving legal standards and regulations, particularly concerning requirements for being a public company. |
These
forward-looking statements are subject to a number of risks, uncertainties and assumptions, including those described in “Risk
Factors” and elsewhere in this prospectus. Moreover, we operate in a very competitive and rapidly changing environment, and
new risks emerge from time to time. It is not possible for us to predict all risks, nor can we assess the impact of all factors on our
business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained
in any forward-looking statements we may make. In light of these risks, uncertainties and assumptions, the forward-looking events and
circumstances discussed in this prospectus may not occur and actual results could differ materially and adversely from those anticipated
or implied in our forward-looking statements.
You
should not rely upon forward-looking statements as predictions of future events. Although we believe that the expectations reflected
in our forward-looking statements are reasonable, we cannot guarantee that the future results, levels of activity, performance or events
and circumstances described in the forward-looking statements will be achieved or occur. Moreover, neither we nor any other person assumes
responsibility for the accuracy and completeness of the forward-looking statements. We undertake no obligation to update publicly any
forward-looking statements for any reason after the date of this prospectus to conform these statements to actual results or to changes
in our expectations, except as required by law.
You
should read this prospectus and the documents that we reference in this prospectus and have filed with the SEC as exhibits to the registration
statement of which this prospectus is a part with the understanding that our actual future results, levels of activity, performance and
events and circumstances may be materially different from what we expect.
TRADEMARKS,
SERVICE MARKS AND TRADE NAMES
We
own or have rights to use a number of registered and common law trademarks, service marks and/or trade names in connection with our business
in the United States and/or in certain foreign jurisdictions.
Solely
for convenience, the trademarks, service marks, logos and trade names referred to in this prospectus are without the ® and ™
symbols, but such references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable
law, our rights or the rights of the applicable licensors to these trademarks, service marks and trade names. This prospectus contains
additional trademarks, service marks and trade names of others, which are the property of their respective owners. All trademarks, service
marks and trade names appearing in this prospectus are, to our knowledge, the property of their respective owners. We do not intend our
use or display of other companies’ trademarks, service marks, copyrights or trade names to imply a relationship with, or endorsement
or sponsorship of us by, any other companies.
USE
OF PROCEEDS
We
estimate that the net proceeds from our issuance and sale of 3,000,000 shares of our common stock (and/or Pre-Funded Warrants
to purchase up to 3,000,000 shares of common stock in lieu thereof) in this offering, at the assumed public offering price
of $2.05 per share, the last reported sale price of our common stock on The NYSE American on January 18, 2024, will be
approximately $5,127,250 (or $5,971,338 if the underwriters exercise in full their option to purchase additional shares
and/or Pre-Funded Warrants), and after deducting the estimated underwriting discounts and commissions and estimated offering expenses
payable by us.
We
intend to use the net proceeds from this offering, along with our existing cash and cash equivalents, as follows:
| |
● |
approximately
$1.2 million to fund clinical trials and product development, including $0.9 million for the ATR-12 Netherton syndrome
program and $0.3 million for the ATR-04 EGFR inhibitor-associated rash program; |
| |
|
|
| |
● |
approximately
$0.5 million for research and development focusing on new product development; |
| |
|
|
| |
● |
approximately
$0.6 million for clinical manufacturing; and |
| |
|
|
| |
● |
the
balance for other general corporate purposes, including in-licensing and partnering activities, laboratory facility improvements,
general and administrative expenses and working capital. |
Our
expected use of net proceeds from this offering represents our current intentions based upon our present plans and business condition.
As of the date of this prospectus, we cannot predict with complete certainty all of the particular uses for the net proceeds to be received
upon the completion of this offering or the actual amounts that we will spend on the uses set forth above. We believe opportunities may
exist from time to time to expand our current business through the acquisition or in-license of complementary product candidates. While
we have no current agreements or plans for any specific acquisitions or in-licenses at this time, we may use a portion of the net proceeds
for these purposes.
The
amounts and timing of our actual expenditures will depend on numerous factors, including the progress of our clinical trials and other
development and commercialization efforts for our initial product candidates, as well as the amount of cash used in our operations. However,
we cannot estimate with certainty the amount of net proceeds to be used for the purposes described above. We may find it necessary or
advisable to use the net proceeds for other purposes, and we will have broad discretion in the application of the net proceeds. Pending
the uses described above, we plan to invest the net proceeds from this offering in short-and intermediate-term, interest-bearing obligations,
investment-grade instruments, certificates of deposit or direct or guaranteed obligations of the government.
DIVIDEND
POLICY
We
have never declared or paid any cash dividends on our common stock and we do not anticipate paying any cash dividends on our common stock
in the foreseeable future. Investors should not purchase our common stock with the expectation of receiving cash dividends. The payment
of dividends on our common stock, if any, in the future is within the discretion of our Board and will depend on our earnings, capital
requirements and financial condition and other relevant facts. We currently intend to retain all future earnings, if any, to finance
the development and growth of our business.
CAPITALIZATION
The
following table sets forth our cash and capitalization as of September 30, 2023:
| |
● |
on
an actual basis; and |
| |
● |
on
a as adjusted basis to reflect our sale of 3,000,000 shares of common stock in this offering at the assumed public offering
price of $2.05 per share, the last reported sale price of our common stock on The NYSE American on January 18, 2024, and
no sale of Pre-Funded Warrants, after deducting underwriting discounts and commissions and estimated offering expenses payable
by us. |
You
should read the information in this table together with our financial statements and related notes and “Management’s Discussion
and Analysis of Financial Condition and Results of Operations” appearing elsewhere in this prospectus.
| | |
September
30, 2023 | |
| (in
thousands) | |
Actual | | |
As
Adjusted | |
| Cash and
cash equivalents | |
$ | 4,400 | | |
$ | 9,527 | |
| Common stock, $0.0001
par value, 100,000,000 shares authorized, 12,097,643 shares issued and outstanding, actual; 15,097,643 shares issued
and outstanding, as adjusted | |
| 1,210 | | |
| 1,510 | |
| Additional paid-in capital | |
$ | 51,475 | | |
$ | 56,601 | |
| Accumulated deficit | |
$ | (46,144 | ) | |
$ | (46,144 | ) |
| Total stockholders’
equity | |
$ | 5,332 | | |
$ | 10,459 | |
| Total
capitalization | |
$ | 5,332 | | |
$ | 10,459 | |
DILUTION
If
you invest in our common stock in this offering, your interest will be diluted to the extent of the difference between the amount per
share paid by purchasers of shares of common stock in this offering and the as adjusted net tangible book value per share of common stock
immediately after the completion of this offering.
As
of September 30, 2023, our net tangible book value was approximately $5.3 million, or $0.44 per share of common stock. Our net tangible
book value per share represents the amount of our total tangible assets reduced by the amount of our total liabilities and divided by
the total number of shares of our common stock outstanding as of September 30, 2023.
After
giving effect to our sale in this offering of shares of our common stock, at the assumed public offering price of $2.05
per share, the last reported sale price of our common stock on The NYSE American on January 18, 2024, after deducting underwriting
discounts and commissions and estimated offering expenses payable by us, and assuming no sale of Pre-Funded Warrants, our as adjusted
net tangible book value as of September 30, 2023 would have been approximately $10.4 million, or $0.69 per share of our
common stock. This represents an immediate increase in as adjusted net tangible book value of $0.25 per share to our existing
stockholders and an immediate dilution of $1.36 per share to investors purchasing shares in this offering.
The
following table illustrates this dilution:
| Assumed public
offering price per share | |
| | | |
$ | 2.05 | |
| Net tangible book value
per share as of September 30, 2023, before giving effect to this offering | |
$ | 0.44 | | |
| | |
| Increase
in net tangible book value per share attributable to new investors purchasing shares in this offering | |
$ | 0.25 | | |
| | |
| As
adjusted net tangible book value per share, after giving effect to this offering | |
| | | |
$ | 0.69 | |
| Dilution per share to
new investors purchasing shares in this offering | |
| | | |
$ | 1.36 | |
The
foregoing tables and calculations are based on 12,097,643 shares of our common stock outstanding as of January 12, 2024 and excludes:
| |
● |
1,288,255
shares of our common stock issuable upon exercise
of outstanding options, with a weighted average exercise price of $1.36 per share, granted pursuant to our 2016 Plan and
2023 Plan; |
| |
● |
approximately
323,736 shares of our common stock issuable upon exercise of outstanding warrants, with a weighted average exercise price of $4.75
per share; |
| |
● |
up
to 450,000 shares of our common stock (and/or Pre-Funded Warrants to purchase up to 450,000 shares of our common stock
in lieu thereof) issuable pursuant to the underwriters’ over-allotment option; |
| |
● |
120,000 shares
issuable upon exercise of a warrant to be issued to the underwriter as part of its compensation in connection with this offering
(up to 138,000 shares if the over-allotment option is exercised in full) at an exercise price of $2.56 per share (125%
of the assumed public offering price per share of the shares of common stock sold in this offering); and |
| |
● |
242,345
shares of our common stock reserved for future grants under our 2016 Plan and 1,960,000 shares of our common stock reserved for future
grants under our 2023 Plan. |
MANAGEMENT’S
DISCUSSION AND ANALYSIS
OF
FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You
should read the following discussion together with our financial statements and the related notes included elsewhere in this prospectus.
This discussion contains forward-looking statements that are based on our current expectations, estimates and projections about our business
and operations. Our actual results may differ materially from those currently anticipated and expressed in such forward-looking statements
as a result of a number of factors, including those which we discuss under “Risk Factors” and elsewhere in this prospectus.
See the section titled “Cautionary Note Regarding Forward-Looking Statements.”
Overview
We
were formed in January 2014 as a biopharmaceutical company focused on developing innovative therapies for precision dermatology using
engineered proteins and live biotherapeutic products. We have built a proprietary platform that includes a microbial library comprised
of approximately 1,500 unique bacterial strains that can be screened for unique therapeutic characteristics. The platform is augmented
by an artificial intelligence and machine learning technology that analyzes, predicts and helps screen our library of strains for drug-like
molecules. The platform also utilizes a licensed genetic engineering technology, which can enable the transformation of previously genetically
intractable strains. We are an early-stage clinical biopharmaceutical company and have not commenced commercial operations.
To
date, we have capitalized our operations primarily through a series of private placements of our convertible preferred stock and convertible
promissory notes and our initial public offering, or IPO, of common stock which closed on June 21, 2023. In connection with our IPO,
we issued 1.5 million shares of our common stock at a public offering price of $5 per share. Concurrent with the close of our IPO, all
of our outstanding shares of convertible preferred stock and convertible promissory notes converted into a total of 8,951,526 shares
of our common stock. As of November 14, 2023, we had 12,097,643 shares of our common stock issued and outstanding. Except as otherwise
indicated, all share and share price in this prospectus gives effect to a forward stock split effected on May 17, 2023 at a ratio of
7.1-for-1.
Results
of Operations
We
are an early-stage clinical biopharmaceutical company, formed in January 2014, and have limited operating history. We have not commenced
revenue-producing operations apart from limited service revenue derived through our JDA with Bayer. Under the terms of the JDA, we are
responsible for testing our library of microbial strains and their natural products for key preclinical properties and Bayer reimburses
us for our development costs. To date, our operations have consisted of the development of our proprietary microbial library, the identification,
characterization and testing of certain bacterial species from our microbial library that we believe are capable of being engineered
to provide significant therapeutic effect and the development of our initial product candidates.
Nine
Months Ended September 30, 2023 Compared to Nine Months Ended September 30, 2022
| | |
Nine
Months Ended September 30, | |
| | |
2023 | | |
2022 | | |
$
Change | | |
%
Change | |
| Service
revenue - related party | |
$ | 596,000 | | |
$ | 253,500 | | |
$ | 342,500 | | |
| 135 | % |
| Total revenue | |
| 596,000 | | |
| 253,500 | | |
| 342,500 | | |
| 135 | % |
| | |
| | | |
| | | |
| | | |
| | |
| Operating expenses: | |
| | | |
| | | |
| | | |
| | |
| General and administrative | |
| 3,443,559 | | |
| 2,583,818 | | |
| 859,741 | | |
| 33 | % |
| Research
and development | |
| 2,188,795 | | |
| 4,425,195 | | |
| (2,236,400 | ) | |
| (51 | )% |
| Total operating expenses | |
| 5,632,354 | | |
| 7,009,013 | | |
| (1,376,659 | ) | |
| (20 | )% |
| | |
| | | |
| | | |
| | | |
| | |
| Loss from operations | |
| (5,036,354 | ) | |
| (6,755,513 | ) | |
| 1,719,159 | | |
| (25 | )% |
| | |
| | | |
| | | |
| | | |
| | |
| Other income (expense): | |
| | | |
| | | |
| | | |
| | |
| Interest income | |
| 1,184 | | |
| 4,056 | | |
| (2,872 | ) | |
| (71 | )% |
| Interest expense | |
| (166,729 | ) | |
| (66,781 | ) | |
| (99,948 | ) | |
| 150 | % |
| Employee retention credit | |
| - | | |
| 229,813 | | |
| (229,813 | ) | |
| (100 | )% |
| Forgiveness of accounts
payable | |
| 56,285 | | |
| - | | |
| 56,285 | | |
| 100 | % |
| Change in fair value of
convertible note | |
| (3,630,100 | ) | |
| - | | |
| (3,630,100 | ) | |
| 100 | % |
| Other
expense | |
| (54,282 | ) | |
| (45,365 | ) | |
| (8,917 | ) | |
| 20 | % |
| Total other income (expense) | |
| (3,793,642 | ) | |
| 121,723 | | |
| (3,915,365 | ) | |
| (3,217 | )% |
| | |
| | | |
| | | |
| | | |
| | |
| Net loss | |
| (8,829,996 | ) | |
| (6,633,790 | ) | |
| (2,196,206 | ) | |
| 33 | % |
| Dividends on preferred
stock | |
| (1,355,347 | ) | |
| (2,076,737 | ) | |
| 721,390 | | |
| (35 | )% |
| Net loss attributable
to common shareholders | |
$ | (10,185,343 | ) | |
$ | (8,710,527 | ) | |
$ | (1,474,816 | ) | |
| 17 | % |
Service
Revenue - Related Party
We
generated $596,000 of service revenue under the Bayer JDA during the first nine months of fiscal 2023 compared to service revenue of
$253,500 under the JDA for the comparable period in fiscal 2022. The increase of $342,500 in service revenue is attributable to an increase
in the amount of reimbursable development costs in 2023.
General
and Administrative
General
and administrative costs during the first nine months of fiscal 2023 increased by $859,741, or 33%, to $3,443,559 from the prior year
period. The increase was primarily related to an increase of $1,167,000 in accounting, financing, legal and insurance costs offset by
a decrease of $381,000 in payroll and related costs and debt issuance costs, and a net increase of $73,741 in other overhead expenses.
Research
and Development
Research
and development expenses include salaries and benefits of all research personnel, payments to contract research organizations, payments
to research consultants, and the purchase of lab supplies. These expenses are offset by income earned from government grant payments.
We generate grant revenue on contracts with various federal agencies and nonprofit research institutions for general research conducted
by us. These grant arrangements also do not meet the criteria for revenue recognition, and amounts earned under these grant contracts
are recorded as a negative research and development expense.
During
the first nine months of fiscal 2023, research and development expenses decreased by $2,236,400, or 51%, to $2,188,795 from the prior
year period. The decrease was primarily related to a decrease of $1,547,000 in research and development related costs attributable to
our efforts in moving our Netherton program forward, a net decrease in payroll and related costs of $702,000 attributable to a reduction
in staff and a net decrease of $12,600 of other costs. There was no government and nonprofit grant revenue received by us during the
first nine months of fiscal 2023 or 2022.
We
expect our research and development expenses to significantly increase in the future primarily due to our planned clinical trial activity
and continued development of product candidates.
Other
Income (Expense)
Our
other income (expense) consists of refundable research and development credits, valuation of warrants, amortization of debt issuance
costs, forgiveness of accounts payable, loss on disposal of equipment, loss on foreign currency translation, employee retention credit,
change in fair value of the convertible note and interest expense. During the first nine months of fiscal 2023, other income (expense)
increased by $3,915,365, or 3,217%, compared to the comparable period in fiscal 2022. The increase was primarily related to an increase
of $3,630,100 attributable to the change in fair value of the convertible note, an increase of $99,948 attributable to interest expense,
an increase of $229,813 attributable to the employee retention credit, a decrease of $56,285 attributable to forgiveness of accounts
payable and a net increase of $11,789 attributable to other income and expense.
Year
Ended December 31, 2022 Compared to Year Ended December 31, 2021
| | |
Years
ended December 31, | |
| | |
2022 | | |
2021 | | |
$
Change | | |
%
Change | |
| Service revenue – related
party | |
$ | 284,000 | | |
$ | 110,000 | | |
$ | 174,000 | | |
| 158 | % |
| Total revenue | |
| 284,000 | | |
| 110,000 | | |
| 174,000 | | |
| 158 | % |
| Operating expenses: | |
| | | |
| | | |
| | | |
| | |
| General and administrative | |
| 3,639,666 | | |
| 3,951,352 | | |
| (311,686 | ) | |
| (8 | )% |
| Research and development | |
| 6,097,938 | | |
| 5,380,102 | | |
| 717,836 | | |
| 13 | % |
| Total operating expenses | |
| 9,737,604 | | |
| 9,331,454 | | |
| 406,150 | | |
| 5 | % |
| Loss from operations | |
| (9,453,604 | ) | |
| (9,221,454 | ) | |
| (232,150 | ) | |
| 3 | % |
| Other income (expense): | |
| | | |
| | | |
| | | |
| | |
| Interest income | |
| 4,818 | | |
| 8,759 | | |
| (3,941 | ) | |
| (45 | )% |
| Interest expense | |
| (251,891 | ) | |
| (66,968 | ) | |
| (184,923 | ) | |
| 276 | % |
| Other income | |
| 65,849 | | |
| 112,141 | | |
| (46,292 | ) | |
| (41 | )% |
| Employee retention credit | |
| 229,813 | | |
| – | | |
| 229,813 | | |
| 100 | % |
| Forgiveness of Payroll
Protection Program loan | |
| – | | |
| 232,506 | | |
| (232,506 | ) | |
| (100 | )% |
| Change in fair value of
convertible note | |
| (1,250,000 | ) | |
| – | | |
| (1,250,000 | ) | |
| 100 | % |
| Other expense | |
| (25,351 | ) | |
| (4,659 | ) | |
| (20,692 | ) | |
| 444 | % |
| Total other income (expense) | |
| (1,226,762 | ) | |
| 281,779 | | |
| (1,508,541 | ) | |
| 734 | % |
| Net loss before income taxes | |
| (10,680,366 | ) | |
| (8,939,675 | ) | |
| (1,740,691 | ) | |
| 19 | % |
| Income tax benefit (expense) | |
| – | | |
| – | | |
| | | |
| | |
| Net loss | |
$ | (10,680,366 | ) | |
$ | (8,939,675 | ) | |
| (1,740,691 | ) | |
| 19 | % |
Service
Revenue - Related Party
We
generated $284,000 of service revenue under the Bayer JDA during fiscal 2022 compared to service revenue of $110,000 under the JDA in
fiscal 2021. The increase of $174,000 in service revenue is attributable to an increased amount of reimbursable development costs in
2022.
General
and Administrative
General
and administrative costs during fiscal 2022 decreased by $311,686, or 8%, to $3,639,666 from the prior year period. The decrease was
primarily related to a decrease of $961,000 in payroll and related costs attributable to the discontinuation of separation benefits paid
to our former chief executive officer and chief operating officer and the reduction in recruiting expenses for our new chief executive
officer, offset by an increase of $607,000 in accounting and legal fees, and $43,000 net increase of other overhead expenses.
Research
and Development
Research
and development expenses include salaries and benefits of all research personnel, payments to contract research organizations, payments
to research consultants, and the purchase of lab supplies. These expenses are offset by income earned from government grant payments.
We generate grant revenue on contracts with various federal agencies and nonprofit research institutions for general research conducted
by us. These grant arrangements also do not meet the criteria for revenue recognition and amounts earned under these grant contracts
are recorded as a negative research and development expense.
During
fiscal 2022, research and development expenses increased by $717,836, or 13%, to $6,097,938 from the prior year period. The increase
was primarily related to an increase of $808,000 in research and development related costs attributable to our efforts in moving our
Netherton program forward offset by a net decrease in payroll and related costs of $85,738 attributable to a reduction in staff. Research
and development expenses in fiscal 2022 and 2021 were offset by $4,426 and $202,509, respectively, of government and nonprofit grant
revenue received by us.
We
expect our research and development expenses to significantly increase in the future primarily due to our planned clinical trial activity
and continued development of product candidates.
Other
Income (Expense)
Our
other income (expense) consists of refundable research and development credits, employee retention credit, forgiveness of the payroll
protection loan, valuation of warrants, amortization of debt issuance costs, change in fair value of the convertible note and interest
expense on the placement of the $1 million unsecured promissory note in January 2021. During fiscal 2022, other income (expense) increased
by $1,508,541, or 734%, compared to fiscal 2021. The increase was primarily related to an increase of $1,250,000 attributable to the
change in fair value of the convertible note, an increase of $229,813 attributable to the employee retention credit, an increase of $184,923
attributable to interest expense offset by a net decrease of $156,195 of other income and expenses.
Liquidity
and Financial Condition
Overview
As
of September 30, 2023, we had total assets of $7.5 million and working capital of $3.4 million. As of September 30, 2023, our liquidity
included approximately $4.4 million of cash and cash equivalents. As of the date of this prospectus, our projected working capital needs
consist of funds with which to further clinical trials and product development, research and development, clinical manufacturing as well
as for other general corporate purposes, including general and administrative expenses. See the section titled “Use of Proceeds.”
Funding
Requirements
We
believe that net proceeds of this offering, along with our cash on hand as of the date of this prospectus, may not be sufficient
to cover our proposed plan of operations over, at least, the 12 months following this offering. We intend to seek additional funds through various financing sources, including
the sale of our equity and debt securities, licensing fees for our technology and joint ventures with industry partners. In addition,
we will consider alternatives to our current business plan that may enable us to achieve revenue producing operations and meaningful
commercial success with a smaller amount of capital. However, there can be no guarantees that such funds will be available on commercially
reasonable terms, if at all. If such financing is not available on satisfactory terms, we may be unable to further pursue our business
plan and we may be unable to continue operations.
To
the extent that we raise additional capital through the sale of equity or convertible debt securities, our common stockholders’
ownership interests will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect
your rights as a common stockholder. Debt financing and preferred equity financing, if available, may involve agreements that include
covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making acquisitions or capital
expenditures or declaring dividends. If we raise additional funds through collaborations, strategic alliances or marketing, distribution
or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams,
research programs or product candidates, or grant licenses on terms that may not be favorable to us. If we are unable to raise additional
funds through equity or debt financings or other arrangements when needed, we may be required to delay, limit, reduce or terminate our
research, product development or future commercialization efforts, or grant rights to develop and market product candidates that we would
otherwise prefer to develop and market ourselves.
The
report of our independent registered public accounting firm for the nine months ended September 30, 2023 states that due to our accumulated
deficit, recurring and negative cash flow from operations there is substantial doubt about our ability to continue as a going concern.
Our financial statements, in Note 1, include disclosure with respect to a substantial doubt about our ability to continue as a going
concern and the report of our independent auditor includes an explanatory paragraph with respect to that substantial doubt.
Contractual
Obligations
Material
contractual obligations arising in the normal course of business primarily consist of operating leases. See Note 15 to our audited financial
statements for future minimum payments under non-cancelable operating leases with initial or remaining terms in excess of one year during
over the next five years.
Cash
Flows
The
following table shows a summary of our cash flows for the periods indicated:
| | |
Nine
Months Ended September 30, | |
| | |
2023 | | |
2022 | |
| | |
| | |
| |
| Net cash used in operating activities | |
$ | (4,778,962 | ) | |
$ | (6,176,777 | ) |
| Net cash used in investing activities | |
$ | (258,274 | ) | |
$ | (239,970 | ) |
| Net cash provided by financing activities | |
$ | 5,944,907 | | |
$ | 4,310,807 | |
| Net increase (decrease) in cash | |
$ | 907,671 | | |
$ | (2,105,940 | ) |
Operating
Activities. During the first nine months of fiscal 2023, operating activities used $4.8 million of cash primarily driven by our net
loss of $8.8 million offset by non-cash items of $4.0 million. During the comparable period of fiscal 2022, operating activities used
$6.2 million of cash primarily driven by our net loss of $6.6 million offset by non-cash items of $400,000.
Investing
Activities. During the first nine months of fiscal 2023, investing activities used $258,274 of cash primarily driven by $235,000
of trademark and patent costs and $23,000 for the purchase of furniture and equipment. During the comparable period of fiscal 2022, investing
activities used $240,000 of cash primarily driven by $221,000 of trademark and patent costs and $19,000 for the purchase of furniture
and equipment.
Financing
Activities. During the first nine months of fiscal 2023, financing activities provided $5.9 million in cash driven by proceeds from
our IPO. During the comparable period of fiscal 2022, financing activities provided $4.3 million in cash primarily driven by the proceeds
from the convertible notes.
Year
Ended December 31, 2022 Compared to Year Ended December 31, 2021
| | |
Years
Ended December 31, | |
| | |
2022 | | |
2021 | |
| Cash Used in Operating Activities | |
$ | (8,349,469 | ) | |
$ | (8,067,359 | ) |
| Cash Used in Investing Activities | |
$ | (336,761 | ) | |
$ | (652,275 | ) |
| Cash provided by Financing Activities | |
$ | 4,134,624 | | |
$ | 992,862 | |
| Net Decrease in Cash and Cash Equivalents | |
$ | (4,551,606 | ) | |
$ | (7,726,772 | ) |
Operating
Activities. During the year ended December 31, 2022, operating activities used $8.3 million of cash primarily driven by our net loss
of $10.7 million offset by non-cash items of $2.4 million. During the year ended December 31, 2021, operating activities used $8.1 million
of cash primarily driven by our net loss of $8.9 million offset by non-cash items of $0.8 million.
Investing
Activities. During the year ended December 31, 2022, investing activities used $0.336 million of cash primarily driven by $0.308
million of trademark and patent costs and $0.028 million for the purchase of furniture and equipment. During the year ended December
31, 2021, investing activities used $0.652 million of cash primarily driven by $0.206 million of trademark and patent costs and $0.446
million for the purchase of furniture and equipment.
Financing
Activities. During the year ended December 31, 2022, financing activities provided $4.1 million in cash primarily driven by the issuance
of a $4.4 million convertible promissory note offset by $0.3 million of deferred offering costs. During the year ended December 31, 2021,
financing activities provided $0.993 million in cash primarily driven by the issuance of a $1 million convertible promissory note.
Critical
Accounting Policies
This
management’s discussion and analysis of our financial condition and results of operations is based on our financial statements,
which have been prepared in accordance with U.S. GAAP. The preparation of these financial statements requires us to make estimates and
judgments that affect the reported amounts of assets, liabilities, and expenses and the disclosure of contingent assets and liabilities
in our financial statements. On an ongoing basis, we evaluate our estimates and judgments, including those related to prepaid/accrued
research and development expenses, share-based compensation and fair value of convertible promissory notes. We base our estimates on
historical experience, known trends and events, and various other factors that are believed to be reasonable under the circumstances,
the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent
from other sources. Actual results may differ from these estimates under different assumptions or conditions.
While
our significant accounting policies are described in more detail in Note 2 to our audited financial statements included elsewhere in
this prospectus, we believe the following accounting policies are the most critical to the judgments and estimates used in the preparation
of our financial statements.
Internal
Control Over Financial Reporting
Internal
control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting
and the preparation of financial statements in accordance with U.S. GAAP. Under standards established by the Public Company Accounting
Oversight Board, or PCAOB, a deficiency in internal control over financial reporting exists when the design or operation of a control
does not allow management or personnel, in the normal course of performing their assigned functions, to prevent or detect misstatements
on a timely basis. The PCAOB defines a material weakness as a deficiency, or combination of deficiencies, in internal control over financial
reporting, such that there is a reasonable possibility that a material misstatement of annual or interim financial statements will not
be prevented, or detected and corrected, on a timely basis.
During
the preparation of our financial statements for the years ended December 31, 2022 and 2021, we and our independent registered public
accounting firm identified a material weakness as it relates to a lack of adequate segregation of accounting functions and the appropriate
accounting for certain warrants in 2021 that were issued in connection with our previously issued but no longer outstanding debt instruments.
We are in the process of implementing measures designed to improve our internal control over financial reporting and remediate this material
weakness. We intend to increase staffing within our accounting infrastructure sufficient to facilitate proper segregation of accounting
functions and to enable appropriate review of our internally prepared financial statements.
Revenue
Recognition
As
discussed in Note 2 to our audited financial statements included elsewhere in this registration statement, under Accounting Standards
Codification, or ASC, 606, Revenue from Contracts with Customers, an entity recognizes revenue when its customer obtains control of promised
goods or services, in an amount that reflects the consideration which the entity expects to receive in exchange for those goods or services.
To
determine the appropriate amount of revenue to be recognized for arrangements determined to be within the scope of ASC 606, we perform
the following five steps: (i) identification of the contract(s) with the customer, (ii) identification of the promised goods or services
in the contract and determination of whether the promised goods or services are performance obligations, (iii) measurement of the transaction
price, (iv) allocation of the transaction price to the performance obligations, and (v) recognition of revenue when (or as) we satisfy
each performance obligation. We only apply the five-step model to contracts when it is probable that the entity will collect consideration
it is entitled to in exchange for the goods or services it transfers to the customer.
When
optional goods or services are offered, we assess the options to determine whether the options grant the customer a material right. This
determination includes whether the option is priced at an amount that the customer would not have received without entering into the
contract. If we conclude the option conveys a material right, it is accounted for as a separate performance obligation. In identifying
performance obligations in a contract, we identify those promises that are distinct. Promised goods or services are considered distinct
when the customer can benefit from the goods or services on their own, or together with readily available resources, and the goods or
services are separately identifiable from other promises in the contract. If a promise is not distinct, it is combined with other promises
in the contract until the combined group of promises is capable of being distinct.
We
estimate the transaction price based on the amount of consideration we expect to receive for transferring the promised goods or services
in the contract. The consideration may include both fixed consideration and variable consideration. At the inception of each arrangement
that includes variable consideration, we evaluate the amount of the potential payments and the likelihood that the payments will be received.
If it is probable that a significant revenue reversal would not occur, the variable consideration is included in the transaction price.
For contracts that include sales-based royalties for licensed compounds, we recognize revenue at the date when the related sales occur.
Finally, we determine whether the contract contains a significant financing component by analyzing the promised consideration relative
to the standalone selling price of the promised goods and services and the timing of payment relative to the transfer of the promised
goods and services. At each reporting date, we reassess the transaction price and probability of achievement of the performance obligations
and the associated constraints on transaction price. If necessary, we adjust the transaction price, recording a cumulative catch-up based
on progress for the amount that was previously constrained.
Revenue
is recognized when (or as) control of a performance obligation is transferred to the customer. When combined performance obligations
contain a promised license and related services or other promises, management judgment is required to determine the appropriate timing
of revenue recognition. In doing so, we must identify the predominant promise or promises in the contract to determine whether revenue
is recognized at a point in time or over time. If over time, we must determine the appropriate measure of progress. If a license is deemed
to be the predominant promise in a performance obligation, we must determine the nature of the license, whether functional or symbolic
intellectual property, to conclude whether point-in-time or over-time revenue recognition is most appropriate. The determination of functional
or symbolic intellectual property requires an assessment of whether the customer is able to exploit and benefit from the license in its
current condition, or if the utility of the license is dependent on or influenced by our ongoing activities or being associated with
us.
At
each reporting date, we calculate the measure of progress for the performance obligations transferred over time. The calculation generally
uses an input measure based on costs incurred to date relative to estimated total costs to complete the transfer of the performance obligation.
Research
and Development Expenses
Research
and development expenses consist primarily of costs incurred in connection with the development of our product candidates. We expense
research and development costs as incurred.
We
accrue an expense for preclinical studies and clinical trial activities performed by our vendors based upon estimates of the proportion
of work completed. We determine the estimates by reviewing contracts, vendor agreements and purchase orders, and through discussions
with our internal clinical personnel and external service providers as to the progress or stage of completion of trials or services and
the agreed-upon fee to be paid for such services. However, actual costs and timing of clinical trials are highly uncertain, subject to
risks and may change depending upon a number of factors, including our clinical development plan.
We
make estimates of our prepaid/accrued expenses as of each balance sheet date in our financial statements based upon facts and circumstances
known at that time. If the actual timing of the performance of services or the level of effort varies from the estimate, we will adjust
the accrual accordingly. Nonrefundable advance payments for goods and services, including fees for clinical trial expenses, process development
or manufacturing and distribution of clinical supplies that will be used in future research and development activities, are deferred
and recognized as expense in the period that the related goods are consumed, or services are performed.
Share-Based
Compensation
We
measure compensation expense for all share-based awards based on the estimated fair value of the share-based awards on the grant date.
We use the Black-Scholes option pricing model to value our share-based awards. We recognize compensation expense on a straight-line basis
over the requisite service period, which is generally the vesting period of the award. We have not issued awards for which vesting is
subject to market or performance conditions.
The
Black-Scholes option-pricing model requires the use of subjective assumptions that include the expected stock price volatility and the
fair value of the underlying common stock on the date of grant. See Note 10 to our audited financial statements included elsewhere in
this prospectus for information concerning certain of the specific assumptions we used in applying the Black-Scholes option pricing model
to determine the estimated fair value of our awards granted.
Estimating
the Fair Value of Common Stock
We
are required to estimate the fair value of the common stock underlying our share-based awards when performing the fair value calculations
using the Black-Scholes option pricing model. Because our common stock is not currently publicly traded, the fair value of the common
stock underlying our stock options has been approved on each grant date by our Board, with input from management.
The
third-party valuations of our common stocks were performed using methodologies, approaches and assumptions consistent with the American
Institute of Certified Public Accountants, Audit and Accounting Practice Aid Series: Valuation of Privately Held Company Equity Securities
Issued as Compensation. In addition, our Board considered various objective and subjective factors to estimate the estimated fair value
of our common stock, including:
| |
● |
the
prices of our preferred stock sold to outside investors in arm’s length transactions, and the rights, preferences and privileges
of our preferred stock as compared to those of our common stock, including the liquidation preferences of our preferred stock; |
| |
● |
the
estimated value of each security both outstanding and anticipated; |
| |
● |
the
anticipated capital structure, which will directly impact the value of the currently outstanding securities; |
| |
● |
our
results of operations and financial position; |
| |
● |
the
status of our research and development efforts; |
| |
● |
the
composition of, and changes to, our management team and board of directors; |
| |
● |
the
lack of liquidity of our common stock as a private company; |
| |
● |
our
stage of development and business strategy and the material risks related to our business and industry; |
| |
● |
external
market conditions affecting the life sciences and biotechnology industry sectors; |
| |
● |
U.S.
and global economic conditions; |
| |
● |
the
likelihood of achieving a liquidity event for the holders of our common stock, such as IPO or a sale of our company, given prevailing
market conditions; and |
| |
● |
the
market value and volatility of comparable companies. |
In
determining the estimated fair value of our common stock, our Board considered the subjective factors discussed above in conjunction
with the most recent valuations of our common stock that were prepared by an independent third party. An independent valuation specialist
was utilized by our Board when determining the estimated fair value of common stock for the awards granted from October 2017 through
December 2021. Our Board, relying on these third-party valuations, approved valuations of our common stock of $3.39 per share as of October
2017, $6.58 per share as of April 2019, $12.09 per share as of September 2020 and $11.05 per share as of December 2021.
Estimating
the Fair Value of Warrants
We
utilize a Black-Scholes method to value our outstanding warrants at each reporting period, with changes in fair value recognized in the
statement of operations. The estimated fair value of the warrant liabilities is determined using Level 3 inputs. Inherent in a Black-Scholes
model are assumptions related to expected share-price volatility, expected life, risk-free interest rate and dividend yield. We estimate
the volatility of our common stock based on market participant assumptions and matches the volatility used to value our stock options.
The risk-free interest rate is based on the U.S. Treasury zero-coupon yield curve on the grant date for a maturity similar to the expected
remaining life of the warrants. The expected life of our outstanding warrants is assumed to be equivalent to their remaining contractual
term. The dividend rate is based on the historical rate, which the Company anticipates to remain at zero.
Recent
Accounting Pronouncements
See
Note 2 to our audited financial statements found elsewhere in this prospectus for a description of recent accounting pronouncements applicable
to our financial statements.
BUSINESS
Overview
We
are an early-stage clinical biopharmaceutical company focused on developing innovative therapies for precision dermatology using engineered
proteins and topical live biotherapeutic products. We have built a proprietary platform that includes a microbial library comprised of
approximately 1,500 unique bacterial strains that can be screened for unique therapeutic characteristics. The platform is augmented by
an artificial intelligence and machine learning technology that analyzes, predicts and helps screen our library of strains for drug like
molecules. The platform also utilizes a licensed genetic engineering technology, which can enable the transformation of previously genetically
intractable strains. Our initial focus is on the development of genetically engineered strains of Staphylococcus epidermidis, or
S. epidermidis, which we consider to be an optimal therapeutic candidate species for engineering of dermatologic therapies. The
particular species demonstrates a number of well-described properties in the skin. As of the date of this prospectus, we have identified,
among our microbial library, over 60 distinct bacterial species that we believe are capable of being engineered to create living organisms
or engineered proteins with significant therapeutic effect.
We
are a pioneer in genetically engineering bacteria for therapeutic use in dermatology. Our goal is to leverage our platforms and internal
microbial library bacterial strains to create new therapeutics that are either engineered living organisms or engineered proteins or
peptides to treat skin diseases. Our initial focus is on the development of our current product candidates, including:
| |
● |
ATR-12,
a genetically modified strain of S. epidermidis for treating the orphan disease, Netherton syndrome, a chronic and sometimes
fatal disease of the skin estimated to affect approximately one to nine in every 100,000, but its prevalence may be underestimated
due to misdiagnosis caused by similarities to other skin diseases. We received Pediatric Rare Disease Designation for ATR-12 by the
United States Food and Drug Administration, or FDA, in 2019. In December 2022, we submitted an investigational new drug application,
or IND, for a Phase 1b clinical trial of ATR-12 in Netherton syndrome patients, and on January 27, 2023 we received notification
from the FDA that the “study may proceed” with respect to the proposed Phase 1b clinical trial. We have commenced our
Phase 1b clinical trial and expect to enroll our first patient in the first quarter of 2024 and report initial safety results
in the second half of 2024. |
| |
|
|
| |
● |
ATR-04,
a genetically modified strain of S. epidermidis for treating the papulopustular rash experienced by cancer patients undergoing
epidermal growth factor receptor inhibitor, or EGFRi, targeted therapy. We intend to submit an IND for a Phase 1b clinical trial
in certain cancer patients undergoing EGFRi targeted therapy by the first half of 2024. Subject to FDA clearance of our IND, we expect
to commence our Phase 1b clinical trial in the second half of 2024. |
| |
|
|
| |
● |
ATR-01,
an engineered recombinant human filaggrin protein for treating ichthyosis vulgaris, a chronic, xerotic (abnormally dry), scaly skin
disease with an estimated incidence and prevalence of 1 in 250, which suggests a total patient population of 1.3 million in the United
States. We are planning to complete lead optimization and IND-enabling studies in 2024 to support an IND filing target in mid-2025. |
| |
|
|
| |
● |
Two
separate strains of bacterial microbes are being investigated and developed by us and Bayer Consumer Care AG, the consumer products
division of Bayer AG, or Bayer, the international life science company. We entered into a Joint Development Agreement, or JDA, with
Bayer in December 2019. Under the terms of the JDA, we are responsible for testing our library of bacterial strains and their natural
products for key preclinical properties. After screening through hundreds of strains, we and Bayer have selected two particular strains
to move forward into further development. Bayer holds the exclusive option to license the patent rights to these strains. In December
2020, Bayer purchased $8 million of our Series B preferred stock, which converted into 1,449,743 shares of our common stock, representing
approximately 12.0% of our outstanding shares of common stock. |
We
also have established partnerships with teams from Carnegie Mellon University and the Fred Hutchinson Cancer Center, or Fred Hutch, two
of the premier academic centers in the United States. Our collaboration with the Carnegie Mellon based team takes advantage of the power
of whole genome sequencing. This partnership is mining our proprietary library of bacterial strains for novel, drug like peptides and
proteins. The artificial intelligence/machine learning technology developed by this team predicts the molecules made by microbes from
their genetic sequences. The system then compares the predictions to the products actually made through tandem mass spectroscopy and/or
nuclear magnetic resonance imaging to refine future predictions. The predictions can be compared to publicly available 2D and 3D protein
databases to select drug like structures.
We
hold an exclusive, worldwide license from Fred Hutch regarding the use of its patented SyMPL technologies for all fields of genetic engineering,
including to discover, develop and commercialize engineered microbial therapies and microbial-derived peptides and proteins for skin
diseases. We are utilizing our licensed patent rights to build plasmids that in order to make genetic transformations that have never
been previously achieved. Our collaboration with Fred Hutch is led by Dr. Christopher Johnston, an expert in microbial engineering, and
the innovator behind the SyMPL technology.
Beyond
our three lead product candidates and collaboration with Bayer, our goal is to develop a broad portfolio of product candidates focused
on expanding the application of our platforms for precision dermatology. We believe that we have established a unique position in advancing
the development of biologics for precision dermatology.

Our
Business Strategies
We
intend to create a broad portfolio of product candidates for precision dermatology through our development of genetically engineered
proteins selected from our proprietary microbial library of approximately 1,500 unique bacterial strains. Our strategy is as follows:
| |
● |
Build
a sustainable precision dermatology company. Our goal is to build a leading precision dermatology company with a sustainable
pipeline of product candidates. To that end, we are focused on rapidly advancing our current pipeline of live biotherapeutic candidates
while actively developing additional product candidates. Each of our current product candidates are proprietary and subject to pending
patent applications. We expect that most, if not all, genetically engineered product candidates we develop will be eligible for patent
protection. |
| |
|
|
| |
● |
Advance
our lead product candidates, ATR-12 and ATR-04, through clinical trials. We expect to report initial safety results
of our Phase 1b clinical trial for our ATR-12 in Netherton syndrome patients in the second half of 2024 and are currently
planning to commence a Phase 1b trial of our ATR-04 in certain cancer patients undergoing EGFRi therapy in the second half
of 2024. We have a cleared IND for ATR-12 and expect to file an IND for ATR-04 in the first half of 2024. |
| |
● |
Broaden
our platform by selectively exploring strategic partnerships that maximize the potential of our precision dermatology programs.
We intend to maintain significant rights to all of our core technologies and product candidates. However, we will continue to evaluate
partnering opportunities in which a strategic partner could help us to accelerate development of our technologies and product candidates,
provide access to synergistic combinations, or provide expertise that could allow us to expand into the treatment of different types
of skin diseases. We may also broaden the reach of our platform by selectively in-licensing technologies or product candidates. In
addition, we will consider potentially out-licensing certain of our proprietary technologies for indications and industries that
we are not ourselves pursuing. We believe our genetic engineering techniques and technologies have applicability outside of the field
of medicine, including cosmetics and in the generation of clean fuels and bioremediation. |
| |
|
|
| |
● |
Leverage
our academic partnerships. We currently have partnerships with investigators at the Fred Hutchinson Cancer Center, Yale University,
Jackson Laboratory for Genomic Medicine, and Carnegie Mellon University. We have an exclusive license from the Fred Hutchinson Cancer
Center covering DNA technologies, which enable the genetic transformation of strains that have previously been genetically intractable.
Our collaboration with investigators at Carnegie Mellon University builds on an artificial intelligence and machine learning technology
to predict the drug like molecules made by the microbes in our library. We have an ongoing scientific advisory board contract with
Dr. Julia Oh of the Jackson Laboratories and have historically worked with Jackson Laboratories through sponsored research agreements
for mouse experiments. We expect to leverage these partnerships and potentially expand them or form other academic partnerships to
bolster our engineering platforms and expand our research and development pipeline. |
| |
|
|
| |
● |
Experienced
management team and Board of Directors. We are led by Francisco D. Salva, our chief executive officer, and Travis Whitfill,
our co-founder and chief operating officer, who have more than 35 years of combined experience in the management of biotechnology
companies and healthcare investing. Mr. Salva was previously a co-founder of Acerta Pharma, which was sold to AstraZeneca for approximately
$6.3 billion in a staged acquisition beginning in 2016. He also worked on the turnaround of Pharmacyclics, which subsequently sold
to Abbvie for approximately $21 billion in 2015. Before that, Mr. Salva spent almost a decade in life sciences venture capital. Mr.
Whitfill served as associate research scientist and currently serves as assistant professor adjunct at Yale University with appointments
in the Departments of Pediatrics and Emergency Medicine. He spent nearly a decade in venture capital as a partner in a biotech-focused
venture capital fund, Bios Partners. He has led numerous grant-funded projects, holds nearly a dozen patents and has co-authored
over 60 publications. Our board of directors, or Board, is comprised of renowned group of senior executives, scientists and investors
in the biotechnology industry. |
Our
Microbial Library and Microbial Drug Delivery Platform
Commensal
microorganisms reside on either the surface of the body or in the mucosa without harming human health. They act on the host’s immune
system to induce protective responses that prevent colonization and invasion by infectious pathogens, and thereby play a crucial role
in maintaining human health across a number of organ systems, particularly in the skin. Diverse communities of microorganisms populate
the skin, and a square centimeter can contain up to a billion microorganisms. These diverse communities of bacteria, fungi, mites and
viruses can provide protection against disease and form dynamic, yet distinct niches on the skin. Together, they make up the skin microbiome.
Many
genetically driven human diseases are systemically or partially related to the dysfunction of specific proteins that are missing or functionally
inert due to a mutation. Since approximately 1982, the biopharmaceutical industry has been genetically engineering recombinant proteins
in bacterial microorganisms for purposes providing therapies that mimic or support the body’s normally functioning proteins and
peptides. For decades, the vast majority of genetic engineering has been limited to primary E. coli and a handful of other bacterial
species, many of which can become pathogenic, inducing infection. In contrast, we have chosen to focus on S. epidermidis because
of its beneficial effects as a commensal, naturally occurring microbe on the skin. Our goal is to leverage our platform and internal
microbial library of over 60 bacterial species to engineer and deliver commensal skin bacteria directly to the target through the stratum
corneum of the skin. At these deeper levels in the skin, engineered microbes can produce the missing or inert proteins and thereby resolve
the underlying disease cause.
S.
epidermidis and Our Proprietary Microbial Library
S.
epidermidis is a strong therapeutic candidate species due to a number of well-described properties in the skin. S. epidermidis
is a gram-positive bacterium that is ubiquitous in the human skin and mucosal flora. As one of the earliest colonizers of the skin,
S. epidermidis plays an important role in cutaneous immunity and maintaining microbial community homeostasis. S. epidermidis
is known to have a beneficial relationship with its host as a skin commensal. The species has shown inhibition of the pathogenic
strain, Staphylococcus aureus, or S. aureus, as well as the strain Propionibacterium acnes, or P. acnes. S. epidermidis
induces keratinocytes to produce antimicrobial peptides and produces non-inflammatory T cell accumulation of both CD4+ and CD8+ T
cells via immune cell signaling. The T cell responses induce re-epithelization of the skin after injury, accelerating repair and wound
closure. For these reasons, we believe S. epidermidis offers several advantages as a vector for topical delivery of therapeutic
proteins.
In
their 2019 study, Stacy and Belkaid, world-leading experts in the skin microbiome, described S. epidermidis as “a ‘poster
child’ of the skin microbiota to illustrate the remarkable diversity of functions a microbe can exert on skin physiology and health.”
We believe that S. epidermidis has enormous strain diversity that can be exploited for therapeutic purposes. In the 2020 Oh Study,
Julia Oh’s lab reported that 1,482 unique strains of S. epidermidis were present on only five individuals. These strains
had not only significant genetic diversity but also large phenotypic diversity. We believe this large inter-strain variation among S.
epidermidis can be exploited. To that end, we collected samples from healthy volunteers to develop and characterize our own strain
library of S. epidermidis that includes over 900 unique S. epidermidis strains with potential for therapeutic use. We have
used this microbial library to screen against selected properties, including antimicrobial peptide secretion, S. aureus killing,
antibiotic sensitivity, and other therapeutically relevant characteristics. We have also collected other species in our library that
includes roughly 60 different skin commensal species that can also be screened for therapeutic purposes.
Figure
1. Representative Species in Azitra Microbial Library

Predictive
Analysis of Our Microbial Library
The
biopharmaceutical industry has seen success in identifying and isolating thousands of bacterial species. Yet only a relatively few such
species, believed to be less than 20, have been engineered to produce proteins or peptides with therapeutic potential. We have partnered
with Chemia Biosciences, Inc., a research and development group from Carnegie Mellon University. Through our collaboration with Chemia
Biosciences, we are able to use their proprietary genomic and peptidomic artificial intelligence and machine learning system, NRPMiner,
to develop and confirm natural product predictions of the proteins, peptides and small molecules that are generated by our proprietary
bacterial library. These predictions are confirmed via tandem mass spectroscopy or nuclear magnetic resonance. The information is then
fed back into the machine learning algorithm to refine the predictions. It can also be compared to existing 2D and 3D protein databases
to look for structural homology of our products to existing protein and peptide drugs. We believe our collaboration with the Carnegie
Mellon based team provides us with a scalable and modification tolerant way to accelerate therapeutic discoveries within our microbial
library.
The
Delivery of our Microbially Produced Drugs
The
delivery of genetically engineered proteins to the subcutaneous target sites is hindered by the natural barrier and the defenses of the
stratum corneum. This is the skin’s outermost layer, which acts as a barrier that prevents unwanted materials from entering the
body. To address this challenge, we have developed a proprietary process capable of facilitating protein delivery in a manner that bypasses
the normally impenetrable stratum corneum. The strategy utilizes the ability of particular microbes to infiltrate into the deeper layers
of the skin. There, the genetically modified microbes act as miniature factories to produce a therapeutic protein or molecule where it
is needed.
Our
protein delivery capability for treating skin conditions is based on engineering S. epidermidis and other microbes to secrete
proteins for drug delivery into the skin. We believe any number of proteins can be engineered and encoded by our bacteria to be produced
and delivered to the skin to treat a variety of skin conditions. We have also added key proprietary features in its platform to facilitate
protein delivery. A key feature of this system is that it bypasses the normally impenetrable skin barrier, a problem of topical protein
delivery. The skin barrier, composed of the stratum corneum, is sealed by enucleated keratinocytes and formed by numerous structural,
physical, and biochemical properties. Other transdermal delivery challenges arise due to susceptibility of protein to enzymatic digestion
by proteases and solubility and diffusion impediments due the hydrophobic surface and the layers of linked corneocytes comprising the
stratum corneum. We address this issue by leveraging the natural homing of S. epidermidis to layers below the stratum corneum.
In preclinical studies, we have shown that S. epidermidis homes to layers below the stratum corneum and delivers proteins into
the deeper epidermis.
To
expand upon our recombinant protein construction capabilities, we have acquired an exclusive license to proprietary technology that disguises
our genetically engineered DNA sequences to enable the production of proteins in previously intractable bacterial species. The technology
from the Fred Hutchinson Cancer Center or Fred Hutch, expands the universe of bacterial species that can be genetically modified. It
is based upon a restriction modification system-silent SyMPL toolset. The SyMPL technology platform makes human-made DNA invisible to
the bacteria’s defenses. In theory, the method can be applied to any type of bacteria. Our current product candidates do not incorporate
the SyMPL technology platform, but we expect that some or all of our future product candidates will do so.
Virtually
all strains of naturally occurring bacteria have defense mechanisms called restriction modification systems. The four types of restriction
modification systems recognize and defend against insertion of foreign DNA used to code recombinant proteins. Functional genetic engineering
of S. epidermidis (as well as S. aureus) has previously been limited due to the presence of Type I and IV restriction systems
in virtually all strains of these bacterial species. These restriction systems recognize methylated cytosine bases in DNA from standard
clone expansion systems (such as E. coli) and hinder incorporation of foreign DNA in the microbe. S. epidermidis was once
believed to be an “untransformable” strain due to its genetic intractability. However, we have been able to overcome S.
epidermidis’ defenses.
Current
genetic engineering processes add specific modifications to disguise human made DNA to trick the bacterium into thinking the intruder
is a part of its own DNA. This approach often takes considerable time and resources to try to match the right disguise to each particular
recognition motif. In contrast, Fred Hutch’s SyMPL technology platform is a systematic “stealth-by-engineering” approach
to overcome restriction modification defense systems. These restriction modification defense systems protect microbes from foreign DNA
and hinder the vast majority of genetic engineering approaches. The SyMPL technology platform is based on the ability to build minicircle
DNA plasmids which lack any of the target recognition motifs for the microbe’s defense systems to identify. The technology uses
the genome and methylome from a target bacteria’s genomic sequence to identify the restriction modification target motifs. They
are then eliminated from the nucleotide sequence of the genetic tool in silico. The resulting sequence is used to build the restriction
modification, SyMPL tools. These are propagated and then used for genetic transformations. Not only does the “stealth by engineering”
approach enable transformations in genetically intractable bacterial strains, but it has also been shown to drastically increase transformational
efficiency. Proof of principle experiments have shown improvements of over 10,000x in yields of genetically engineered colonies.
In
January 2022, Fred Hutch granted us an exclusive worldwide, royalty bearing license to the patent rights, and a non-exclusive worldwide,
royalty bearing license to the related know-how, for the SyMPL technology platform in all fields of use. For more information related
to the intellectual property acquired pursuant to the Fred Hutch license agreement, see the section titled “Business-Licenses
and Intellectual Property Rights.”
Our
Product Candidates
ATR-12
for the treatment of Netherton syndrome
ATR-12
is our proprietary and patent-pending drug candidate that contains a novel strain of S. epidermidis which has been genetically
modified to express and secrete an active fragment of the full-length protein called the lympho-epithelial Kazal-type related inhibitor,
or LEKTI. It has also been engineered to be auxotrophic, meaning that it requires the D-alanine nutrient in its formulation to survive
and propagate. This provides an additional level of safety against potential systemic infection. ATR-12 is a topical application intended
to address the underlying cause of Netherton syndrome, by replacing deficient LEKTI with an active segment of human recombinant LEKTI,
or rhLEKTI-D6, to counter the dysregulated skin serine protease activity observed in Netherton syndrome patients. The uncontrolled serine
protease activity leads to a profound skin barrier defect and the release of pro-inflammatory and pro-allergic mediators by keratinocytes
and immune cells. As of the date of this prospectus, there is no known therapy for the cure or effective treatment of Netherton syndrome.
We believe ATR-12 has the potential to be the first therapy to effectively treat this disease of the skin. Based on the Barbati and Sun
Studies, we believe that ATR-12 represents a potential $250 million global sales opportunity by mid-2030.
Netherton
syndrome overview
Netherton
syndrome is a rare, autosomal recessive disease estimated to affect approximately one in every 200,000, but its prevalence may be underestimated
due to misdiagnosis. It is a chronic disease of the skin, characterized by severe inflammation, pruritus, scaling, red, and dehydrated
skin. Infants born with Netherton syndrome may suffer from a failure to thrive, and it has been reported that approximately one in ten
infants with Netherton syndrome die in their first year of life. Those that survive face a lifetime of skin disease challenges including
red, scaly skin, hair defects and an ongoing higher than normal risk for infection and allergy.
Netherton
syndrome is caused by mutations in the SPINK5 gene, which codes for the serine protease inhibitor Lympho-epithelial Kazal-type
related inhibitor, or LEKTI. The function of LEKTI is to inhibit enzymes in the epidermis, such as kallikreins 5, 7 and 14, or KLK5,
KLK7 and KLK14, which facilitate the shedding of skin cells in a process known as desquamation. When LEKTI is absent or has reduced activity,
excess shedding results and the skin is sensitive, open, and appears red and scaly. This is accompanied by the detachment of the stratum
corneum, leading to severe barrier dysfunction, dehydration and potential exposure to environmental agents, such as chemicals. Histopathology
and immunofluorescence staining of skin from a Netherton syndrome patient compared to healthy volunteer reveal an absence of LEKTI and
abnormalities in the skin such hyperkeratosis, epidermal thickening, and reduction of the basophilic keratohyalin granules.

Figure
2: Netherton syndrome pathophysiology and LETKI deficiency
Netherton
syndrome can range in severity from mild, such as red patchy areas of the skin, to life threatening. The degree of severity of the disease
correlates directly with the extent of loss of function of LEKTI on the skin. Netherton syndrome appears shortly after birth and is most
severe in the first year of an infant’s life. Survival beyond the first year is common in most cases, but the implications of the
disease are a lifelong challenge.
As
of the date of this prospectus, there is no known cure for Netherton syndrome and treatment options are limited. Dermatologic interventions
to treat the severe skin manifestations of Netherton syndrome include moisturizers, topical corticosteroids, and calcineurin inhibitors,
all of which are limited in that they do not provide sustained remediation. Given the severity of disease during neonatal stages, fluid/electrolyte
and diet support are needed in addition to treating infections that often arise in these patients. While immunoglobulin therapy to address
immunodeficiencies associated with Netherton syndrome has shown limited success, a sustained remediation of skin barrier defects, induced
by dysregulation of LEKTI, is currently unavailable.
Our
solution – ATR-12 for the treatment of Netherton syndrome
ATR-12
is a topical ointment containing an S. epidermidis strain, SE351, that has been genetically modified to express LEKTI from the
chromosome. The SE351 strain has also been engineered to be auxotrophic for D-alanine, which means it cannot survive without the exogenous
D-alanine nutrient provided in the formulation. ATR-12 is intended to address the underlying cause of Netherton syndrome by replacing
deficient/dysfunctional LEKTI with an active, recombinant, human fragment of the full-length protein, rhLEKTI-D6. The treatment consists
of applying ATR-12 to affected areas. rhLEKTI-D6 produced by SE351 will counter the dysregulated skin serine protease activity observed
in Netherton syndrome patients, to restore skin barrier function and reduce inflammation. We believe that among the important advantages
of this approach is the potential to deliver rhLEKTI-D6 over time into the lower layers of the stratum corneum and epidermis, the primary
sites of dysregulation in patients with Netherton syndrome.
The
S. epidermidis strain selected to deliver rhLEKTI-D6 to the skin, SE351, was selected from our proprietary strain collection.
This strain is characterized by low virulence and is a non-biofilm forming host strain. To further enhance the safety of ATR-12, we have
engineered the microbe for D-alanine to be auxotrophic. The key advantage to engineering auxotrophy is the ability to control growth
and halt potential infection. Full length human LETKI, a 15-domain protein (145 kDa), is too large for reliable bacterial expression
and secretion. Given evidence that fragments of the full-length protein are sufficient to counter the dysregulated skin serine protease
activity observed in Netherton syndrome patients, we selected D6 for recombinant expression in S. epidermidis.
In
May 2020, we received Rare Pediatric Disease Designation from the FDA for ATR-12. As a result, if we are able to obtain approval for
ATR-12 from the FDA in pediatrics, we will be eligible to receive a Priority Review Voucher, which can be used by us to obtain FDA review
of a New Drug Application or Biologics License Application for this or another drug candidate in an expedited period of six months. These
vouchers are often transferable, and some have been sold for over $100 million.
Preclinical
data for ATR-12
As
of the date of this prospectus, we have conducted several in vivo and ex vivo experiments that collectively support the
potential efficacy of ATR-12 as a disease modifying therapy for patients with Netherton syndrome. The genetically engineered strain of
S. epidermidis used in the formulated ATR-12 drug product is called SE351. In 2021, we conducted in vitro studies to assess the
ability of exogenously applied SE351 to colonize sterile reconstructed human epidermis. SE351 successfully colonized the reconstructed
human epidermis and, furthermore, no S. epidermidis colonization occurred without D-alanine present, confirming that D-alanine
must be supplied for SE351 growth on skin. These data suggest that SE351 is capable of colonizing human skin, and that colonization can
be controlled with D-alanine supplementation.
Additionally,
in vitro studies using tape stripped skin from healthy volunteers spiked with KLK5 to mimic Netherton syndrome showed that diluted
SE351 culture supernatant dose-dependently inhibited trypsin-like activity (KLK5 activity). Trypsin-like activity in the Netherton syndrome
surrogates returned to normal healthy levels when a solution containing ≥0.5% of the SE351 culture supernatant was added.

Figure
3: In Vitro Netherton Syndrome Model Using Human Skin Tape Strip Extracts Supplemented with Disease Level KLK5 Activity
In
addition, results from an ex vivo pig skin model demonstrate that a single topical dose of ATR-12 at 3 dose levels led to secretion
of active rhLEKTI-D6. Finally, data from an ex vivo healthy human skin model demonstrate that a single topical dose of ATR-12
administered at the maximum intended dose of 109 CFU/g delivers enough active rhLEKTI-D6 into the lower layers of the stratum
corneum to effectively inhibit the protease, kallikrein 5 (“KLK5”), at levels typically observed in patients with Netherton
syndrome.
In
particular, data from an ex vivo healthy human skin model demonstrate that a single topical dose of ATR-12 administered at the
maximum intended dose of 109 Colony Forming Units per gram (CFU/g) delivers enough active rhLEKTI-D6 into the lower layers
of the stratum corneum to effectively inhibit KLK5 at levels typically observed in patients with Netherton syndrome. Amounts of LEKTI
activity in layers extracted were from tape strip samples from ex vivo human skin treated with placebo and ATR-12. The collection
proceeded right after skin application (T = 0 hours, white bars) or after 8 hours incubation at 30°C (T = 8 hours, black bars). Total
LEKTI activity levels were obtained by adding the pmol amounts through layers 1 to 30 of placebo (grey bars) or ATR-12 (black bars) samples.
Data are the average ± a standard deviation (SD) of 3 independent samples (N = 3). Statistical analysis was carried out using
two-way ANOVA, and ** represents p <0.01.

Figure
4: LETKI activity in Placebo and ATR-12-Treated Skin Samples Following 0- and 8-hour Incubation
In
addition, a single therapeutic dose of ATR-12 over 24-hour incubation yielded ~2-fold higher LEKTI activity compared to 8-hour incubation.
This indicates continuous production of functional rhLEKTI-D6 by ATR-12 over time.

Figure
5: LETKI activity in Placebo and ATR-12-Treated Skin Samples Following 24-hour Incubation
In
vitro stoichiometry work performed by Azitra indicates that KLK5 requires 2 molar equivalents on the rhLEKTI-D6 protein for inhibition
(as measured by IC50). Historical studies have indicated that Netherton syndrome patients to show up to ~6 fold the amount
of KLK5 that the amounts found in normal skin. This equates to 60 pmol of KLK5 per given area. The studies shown above indicate that
SE351 delivered 350 pmol of rhLEKTI-D6 at 8 hours and it delivered 700 pmol of rhLEKTI-D6 at 24 hours. This represents a 5- to 11-fold
amount above the predicted amount required for activity.

Figure
6: In vitro stoichiometry of LEKTI-D6 to inhibit KLK5
In
2022, we obtained pre-IND correspondence with the FDA for purposes of discussing our proposed regulatory pathway for ATR-12 and obtaining
guidance from the FDA on the preclinical plan leading to the filing and acceptance of an IND application for ATR-12. In December 2022,
we filed an IND for a first-in-human trial of ATR-12 in Netherton syndrome patients. Our IND proposes a Phase 1b multi-center, randomized,
double-blind, single dose level, placebo-controlled clinical study of ATR-12 in patients with Netherton syndrome. The primary endpoint
is safety and secondary endpoints will include signals of efficacy and pharmacokinetics. Exploratory endpoints include immune and inflammatory
mechanism biomarkers. On January 27, 2023, we received notification from the FDA that the “study may proceed” with respect
to the proposed Phase 1b clinical trial with initial safety results expected in the second half of 2024.
ATR-04
for the Treatment of EGFRi-Associated Rash
ATR-04
is our proprietary and patent-pending drug candidate that contains a novel strain of S. epidermidis, SE484, which has been genetically
modified to be auxotrophic tor D-alanine. ATR-04 is a topical application intended to address the papulopustular rash experienced by
cancer patients undergoing epidermal growth factor receptor inhibitor, or EGFRi, targeted therapy. We believe this product candidate
represents a potential $1 billion global sales opportunity by 2030.
EGFRi-Associated
Rash Overview
Targeted
cancer therapies have produced significant treatment advances for patients diagnosed with a variety of tumor types, but they are also
associated with unique dermatologic toxicities that may hamper treatment efforts and cause significant physical and psychological discomfort
for patients. Prevention and management of these toxicities may allow patients to tolerate treatments better, remain on therapy longer
and thereby potentially receive maximum clinical benefit from the drug. One such class of targeted cancer therapy includes EGFR inhibitors.
EGFR is a protein on the surface of cells that helps them grow and divide. It is also a key factor in certain malignancies, and its activity
enhances tumor growth, invasion, and metastasis. While systemic exposure to EGFRi agents suppresses EGFR at the target cancer site, it
also suppresses EGFR throughout the body. In the skin, EGFR regulates multiple keratinocyte functions including proliferation, adhesion
and migration, survival, and differentiation. Consequently, inhibition of EGFR in the skin results in adverse skin reactions, which make
it difficult for patients to stay on these effective therapies.
Dermatologic
toxicities are amongst the most prevalent side effects seen with EGFRi-targeted therapies. The papulopustular rash is the earliest and
most common dermatologic adverse event of EGFRi treatment, often occurring in 50-80% of patients, depending on the drug, the cancer being
treated, and the treatment regimen. The appearance of the papulopustular rash is a dose-dependent skin drug reaction, which usually develops
in the first one to two weeks and peaks at three to four weeks on therapy. The intensity of the rash may start to decrease after two
weeks but can persists over the entire course of EGFRi treatment. The rash is characterized clinically as tender erythematous papules,
which after a few days evolve into pustules and then into crusts on the face, scalp, chest, and upper back. The rash is often accompanied
by severe xerosis and at times serious cutaneous bacterial infection, primarily S. aureus. While most skin rash episodes are considered
mild to moderate, some are severe. In many cases the rash leads to severe quality of life issues and can even lead to the interruption
or cessation of the EGFRi treatment.
The
current standard of care for rash treatment in patients undergoing EGFRi treatment varies depending on the rash severity. Typically,
skin moisturizers, topical steroids and doxycycline are administered prophylactically from the start of EGFRi therapy and are continued
throughout the entire treatment period. If the rash continues to advance, oral steroids and/or antibiotics are administered. However,
there are known systemic adverse events associated with these adjunctive therapies, and we believe that physicians and patients try to
limit their use. In addition, research indicates that oral antibiotics lead to a disruption in the gut microbiome, which in turn leads
to a decrease in the effectiveness of targeted therapies, including EGFRi. Given the high incidence rate of rash that continues with
these patients, as well as the concerns related to potential impacts of antibiotics on these therapies, we believe there is a clear unmet
medical need for additional safe and effective adjunctive therapies for addressing papulopustular skin rash.
Based
on studies conducted by Satoh and Lichtenberger, the cytokine, Interleukin-36 gamma, or IL-36γ, and S. aureus are linked
to and play a significant role in the rashes experienced by patients treated with EGFRis. IL-36γ, is elevated in the skin of patients
undergoing EGFRi therapy. In 2020, Satoh used gene expression profiling to identify IL-36γ as a candidate driver of EGFRi/MEKi
skin toxicity. It is induced by EGFR inhibition and Cutibacterium acnes that synergistically induce IL-36γ in the skin and
subsequently IL-8 and NF-κB, which leads to cutaneous neutrophilia. IL-36γ could be a key therapeutic target in treating
EGFRi-induced rashes. In 2013, Lichtenberger noted high rates (70%) of bacterial infection in patients (n=107) on EGFRi and proposed
a mechanism of EGFR ablation leading to S. aureus-induced infection in mice. The study noted a majority of the patients were positive
with S. aureus (54%). Mechanistically, the authors noted that EGFRi therapy impairs host defense: impaired expression of antimicrobial
peptides, especially against S. aureus; and lowered expression of tight junctions. Also, the study revealed EGFR ablation leads
to skin barrier defects as well as impaired cutaneous immune response and cytokine expression.
Our
solution – ATR-04 for the treatment of EGFRi-associated rash
ATR-04
is our formulated, drug product candidate for the treatment of EGFRi associated rash. It includes a novel auxotrophic strain of S.
epidermidis strain that was selected from our microbial strain library, based on desired properties of IL-36γ reduction and
inhibition of S. aureus and its biofilms. The current lead strain is called SE484. We then genetically engineered SE484 to be
auxotrophic tor D-alanine and to create our drug product candidate, ATR-04.
SE484
was chosen from our microbial library based on key characteristics such as inhibition of IL-36γ as well as its effect against S.
aureus. Together, we expect these mechanisms of action to lead to significant reductions in rash severity among patients undergoing
EGFRi therapy.
We
believe that ATR-04 has the potential to address current limitations to treatment of EGFRi-associated rash:
| |
● |
Reduced
antibiotic use. From our surveys of clinicians and key opinion leaders, practitioners are reluctant to prescribe systemic antibiotics
to patients undergoing EGFRi therapy. These patients can often be prescribed antibiotics for more than 12 months and suffer from
antibiotic-related adverse events. We believe ATR-04 would reduce the need for antibiotics in these patients and lead to fewer adverse
events due to EGFRi and antibiotic use. |
| |
|
|
| |
● |
Better
EGFRi compliance. Up to 20% of patients undergoing EGFRi therapy discontinue due to adverse events, primarily due to rashes.
We believe we can reduce discontinuation rate in patients undergoing EGFRi therapy and thus increase compliance. |
| |
|
|
| |
● |
Higher
quality of life. Many patients on EGFRi therapy report a poor quality of life due to adverse events and papulopustular rashes.
Current treatment options fail to adequately reduce these adverse events. We believe ATR-04 therapy in patients undergoing EGFRi
therapy will have reduced rash severity and thus a higher quality of life. |
Preclinical
data of ATR-04
We
screened over 100 strains based on safety (e.g., lack of antibiotic resistance) and biological activity (e.g., IL-36γ inhibition
and activity versus S. aureus) and designated SE484 as our lead candidate strain. After engineering this strain to be auxotrophic
for D-alanine, we nominated this candidate for use as the active microbe in the ATR-04 drug product formulation.
EGFRi-associated
rash is a condition that is characterized by redness, itchiness, and irritation of the skin, and is induced by certain cancer treatments.
It was shown, by gene expression profiling (Satoh et al 2020), that skin biopsy samples from patients suffering from EGFRi-associated
rash had elevated levels of cytokines IL-36γ (IL-36 gamma) and IL-8 compared to skin from healthy donors. These are proinflammatory
cytokines that are signaling molecules of the immune system that increase the intensity of an immune response and can cause tissue damage.
In addition to elevated cytokine levels, EGFRi-treated patients have impaired skin barrier function. Infection with pathogenic strains
of S. aureus exacerbates the EGFRi-induced cutaneous disease.
Our
work was focused on identifying a Staphylococcus epidermidis strain, a skin commensal, that reduces IL-36γ levels and thus
reduces the rash associated with EGFRi. We reasoned that many species of bacteria that live on human skin probably survive there because
they have evolved ways to reduce the human immune system’s response to their presence, and we might be able to identify a resident
human skin commensal bacteria that survives thereby specifically reducing IL-36γ activity.
To
identify such a Staphylococcus epidermidis strain, we developed an in vitro assay to measure the levels of IL-36γ
and IL-8 that are produced by human skin cells that are grown in culture. The cell line we used is called HaCaT and is derived from human
keratinocytes, which are a cell type in the epidermis. In order to simulate the inflammatory phenotype of EGFRi-related disease of the
skin, HaCaT cells were stimulated with an immunostimulant, polyinosinic:polycytidylic acid, or poly I:C, which causes them to secrete
elevated levels of IL-36γ and IL-8. This assay was used to identify and evaluate the ability of different S. epidermidis
strains to lower IL-36γ and IL-8 levels.
We
screened over 100 strains based on safety (e.g., lack of antibiotic resistance) and biological activity (IL-36γ inhibition and
activity against S. aureus) and designated SE484 as our lead candidate strain. After engineering this strain to be auxotrophic
for D-alanine, so that it will grow only if provided with D-alanine, we also eliminated an anti-biotic resistance gene. Then, we nominated
this candidate for use as the active microbe in the ATR-04 drug product formulation.
To
test the ability of SE484 to reduce IL-36γ on a skin-like model, erlotinib was used to induce IL-36γ secretion on reconstructed
human epidermis, or RHE. Simultaneous application of SE484 with erlotinib reduced IL-36γ to a level comparable RHE that had not
been treated with erlotinib, showing that SE484 acts on a skin-like model to reduce this pro-inflammatory cytokine. Figure 7 shows the
results of two experiments to measure IL-36γ reduction by SE484. In Figure 7A, a cell-free supernatant (CFS) from a culture of
SE484 was applied to RHE, while in Figure 7B live cells of SE484 (either 1x108 CFU or 1x109 CFU) were applied to
RHE to measure the ability of SE484 to reduce IL-36γ. In both cases, cell-free supernatant or SE484 cells, erlotinib-induced IL-36γ
levels were reduced.

Figure
7. The anti-IL-36g activity of SE484 on RHE. Reconstructed human epidermis, or RHE, was treated for 72 hours with 1 mM erlotinib
alone or with cell-free supernatant (CFS) from SE484 culture (A), or with approximately 108 or 109 CFU of SE484
(B). RHE feeding media was then assayed by ELISA for IL-36γ levels.

Figure
8. IL-8 induction by Poly I:C is Reduced by CS of SE484. The presence of culture medium from SE484 prevents the stimulation and release
of IL-8 by poly I:C (red arrow). An inhibitor of poly I:C is used as control (blue arrow). Data are representative of two independent
experiments. CS = culture supernatant.
Figure
8 shows the inhibitory effect of SE484 culture supernatant on the induction of IL-8 by poly I:C. Similar to IL-36γ, when poly I:C
is added to HaCaT cells, IL-8 is also secreted, several fold above background (as seen in untreated HaCaT and SE484-treated HaCaT). However,
in the presence of SE484, lower levels of IL-8 were detected, thus further demonstrating the efficacy of SE484 to inhibiting the proinflammatory
pathway involved in EGFRi-related rash.
Our
results show that culture media of S. epidermidis strain SE484, which was isolated from a healthy human volunteer, can reduce
the level of IL-36γ and IL-8 produced by HaCaT cells (Figure 7 and Figure 8, respectively) and thus help in the treatment of EGFRi-
related rash. In addition to its anti-IL-36γ property, SE484 also has broad activity against different methicillin-resistant S.
aureus, or MRSA, strain types as well as methicillin sensitive S. aureus, or MSSA. The ability of SE484 to reduce IL-36γ/IL-8
levels as well as its activity against S. aureus and the engineered D-alanine auxotrophy enabled us to nominate strain SE484 for
use as the active microbe in the ATR-04 drug product formulation to form the basis of a treatment and reduce the severity of EGFRi rash.
We
have also shown that SE484 leads to in vitro inhibition of known virulent strains USA300, which is resistant to methicillin, and
MSSA, which is sensitive to methicillin. The following data show that ATR-04 reduces the ability of the pathogenic S. aureus bacterial
species to grow and instigate infections that are seen in patients with EGFRi rash.

Figure
9. Epidermin-expressing SE484 kills S. aureus with similar activity as mupirocin on in vitro agar plates.
We
are proposing an initial study of SE484 in the ATR-04 formulation in patients. It is contemplated to be a Phase 1b multi-center, randomized,
double-blind, single-dose, placebo-controlled trial in patients with colorectal or head and neck cancer who are initiating EGFRi therapies.
The primary endpoint is safety, and secondary endpoints will include efficacy and Quality of Life, or QoL We are planning to submit an
IND by the first half of 2024. Subject to FDA clearance of our IND, we expect to commence our Phase 1b clinical trial in the second half
of 2024.
ATR-01
for the treatment of ichthyosis vulgaris
ATR-01
is our drug product candidate intended to treat ichthyosis vulgaris. The program is currently investigating a proprietary and patent-pending
novel engineering segment of human filaggrin protein. ATR-01 is being developed as a topical application intended to address ichthyosis
vulgaris, a chronic scaly skin disease with an estimated incidence and prevalence of 1 in 250, which gives a total patient population
of 1.3 million in the United States. Ichthyosis vulgaris is caused by loss-of-function mutations in the gene encoding filaggrin Using
synthetic biology tools for protein engineering, we attached a cell penetrating peptide to filaggrin, which helps facilitate deeper skin
delivery for filaggrin. This is designed to overcome the impenetrability of the skin barrier, which would otherwise limit topical protein
delivery.
Ichthyosis
vulgaris overview
Ichthyosis
vulgaris, or IV, is a chronic, xerotic, scaly skin disease with an estimated incidence and prevalence of 1 in 250, which gives a total
patient population of 1.3 million in the United States. Clinical features of IV usually appear at around 2 months of age and include
generalized xerosis and fine, white to gray scales that are prominent on the abdomen, chest, and extensor surfaces of the extremities.
Although rare, some IV patients also experience hypohidrosis and heat intolerance. The pathogenesis of IV has long been identified as
a decrease in the size or number, or even a complete absence of, epidermal keratohyaline granules. In addition, patients with IV are
at increased risk for atopic dermatitis, asthma and allergies.
Ichthyosis
vulgaris is an autosomal semidominant disease caused by loss-of-function mutations in the gene encoding filaggrin. Filaggrin is an essential
structural protein that is derived from profilaggrin, which breaks down into individual filaggrin units in the stratum corneum. These
reinforce the skin barrier by binding to keratins and other intermediate filament proteins in the keratinocyte cytoskeleton. Many studies
have identified loss-of-function mutations in FLG in IV patients, and these mutations are associated with disorganized keratin
filaments, skin barrier defects and microfractures in the stratum corneum leading to enhanced percutaneous allergen sensitization. Moreover,
filaggrin and its breakdown products have significant additional functions in the skin including moisturizing the skin (via hygroscopic
amino acids or “natural moisturizing factors”), effecting production of antimicrobial molecules (particularly against S.
aureus) and maintaining both a beneficial lipid profile and pH in the skin.
There
are few effective therapies for the treatment of IV. Current treatment options for IV include primarily topical water evaporation suppressants
(e.g., sodium chloride, urea, lactic acid, salicylic acid), and, to a lesser extent, moisturizers (e.g., glycerol, propylene glycol,).
Topical retinoids may also be prescribed in an effort to slow the body’s production of skin cells. However, long-term retinoid
use is not ideal. Of particular concern is the teratogenic effect of all retinoids, which limits their use in women of child-bearing
potential. Chronic toxicities from long term therapy with retinoids may result in skeletal abnormalities. Furthermore, the chronic use
of retinoids in children may inhibit their growth. Notably, many patients with IV experience a significantly reduced quality of life,
due to self-consciousness and social embarrassment, and see a negative impact on domestic life, educational/professional lives and even
leisure/sports activities.
Our
solution – ATR-01 for the treatment of ichthyosis vulgaris
It
is now known that IV is caused by loss-of-function mutations in the gene encoding filaggrin, leading to disorganized keratin filaments,
skin barrier defects and microfractures in the stratum corneum, and resulting in enhanced percutaneous allergen sensitization as well
as bacterial and viral skin infection. We are developing ATR-01 as a novel treatment modality for IV that directly addresses the disease
pathophysiology. ATR-01 consists of FLG9-10 functional unit of the human FLG protein with an attached cell penetrating peptide. The goal
is to supplement the skin with stable delivery of hFLG via topical application and deeper skin penetration with a cell penetrating peptide.
Preclinical
data for ATR-01
Human
FLG units (domains 9-10) were evaluated on human skin explants (from plastic surgery) ex vivo. The skin barrier of the explants
was compromised by repeated tape-stripping such that transepidermal water loss, or TEWL values were significantly increased compared
to normal skin. As shown in the example below, daily topical application of a human filaggrin unit with a cell penetrating peptide for
five days resulted in a dose-dependent (not shown) rapid improvement in TEWL, suggesting improved skin barrier. Thus, topical delivery
of a recombinant hFLG unit coupled with a cell penetrating peptide can improve/accelerate the repair of damaged human skin barrier.

Figure
10: Topical filaggrin application on tape stripped ex vivo human skin following human filaggrin application.
Lastly,
we have shown that topical filaggrin can improve skin barrier defects in filaggrin-deficient mouse models. Recombinant mouse filaggrin,
or mFlg, was applied to the tail of flaky tail, or FT, mice (a mouse model that has a knockout in the filaggrin gene) once daily
for 2 weeks (50 µg total protein/tail sections or 15.2 µg total protein/cm2). Daily treatment with mFlg significantly
improved transepidermal water loss in FT mice when treated (FT+FLG group) compared to vehicle (“baseline” group). The third
group on the X-axis is a normal, control, wild type group (WT) that does not have the filaggrin gene knocked out. Treatment of
damaged mouse skin with recombinant mFlg combined to a cell penetrating peptide improved damaged mouse skin barrier (Figure A below).
Additionally, histological analysis of the epidermis of the mouse tail sections showed tendency for improved stratum corneum thickness
with mFlg treatment (Figure B below). In this graph, the Y-axis represents thickening of the stratum corneum, which starts to fractionate
or scale after growing past a normal thickness. Treatment with mFlg improved the thickness in four of six of the samples.

Other
Potential Product Candidates
Beyond
our three lead product candidates, our goal is to develop a broad portfolio of product candidates focused on expanding the application
of our platforms for precision dermatology. We have a proprietary platform for discovering and developing therapeutic products for precision
dermatology. Our platform is built around a microbial library comprised of approximately 1,500 unique bacterial strains to allow screening
for unique therapeutic characteristics and utilizes microbial genetic technology that analyzes, predicts and engineers the proteins,
peptides and molecules made by skin microbes. Our ability to genetically engineer intractable microbial species is uniquely leveraged
by our exclusive license to the SyMPL technology.
Bayer
Joint Development Agreement
In
December 2019, we entered into a Joint Development Agreement, or JDA, with Bayer pursuant to which we agreed to the joint development
of certain strains selected from our proprietary microbial library. We and Bayer have agreed to cooperate in the identification and in
vitro and ex vivo characterization of microbial strains for topical formulations. Bayer paid us a one-time $150,000 payment
upon execution of the JDA and has agreed to reimburse us for our development costs. In October 2021, Bayer expanded the option agreement
and paid us $375,000 for additional characterization work. We have granted Bayer an option to acquire an exclusive royalty bearing license
for up to six strains subject to development activities under the JDA, including an exclusive royalty bearing license to any related
patent rights. After screening through hundreds of strains, we and Bayer have selected two particular strains to move forward with in
vitro and ex vivo characterization, which we intend to develop as potential over-the-counter cosmetic products. We completed
the characterization work and delivered the data to Bayer in the fourth quarter of 2023, at which time Bayer has 12 months to exercise
its option to license the strains and related patents. Upon the conclusion of the characterization studies and our delivery of the data
to Bayer, the JDA will end, but Bayer has the option to license the strains and related patents. As of the date of this prospectus, we
have not negotiated a commercial license agreement with Bayer and we will not do so until such time, if ever, as Bayer exercises its
option to acquire an exclusive royalty bearing license.
In
September 2020, Bayer’s venture capital group, LEAPS by Bayer, purchased $8 million of our Series B preferred stock.
Sales
and Marketing
Given
our stage of development, we have not yet established a commercial organization or distribution capabilities. We plan to build focused
capabilities in the United States to commercialize our development programs focused on live biotherapeutic products and recombinant proteins
for the treatment of skin diseases, where we believe the patient populations and medical specialists for the indications we are targeting
are sufficiently concentrated to allow us to effectively promote our products, if approved for commercial sale, with a targeted sales
team. In other markets for which commercialization may be less capital efficient for us, we may selectively pursue strategic collaborations
with third parties in order to maximize the commercial potential of our product candidates.
Manufacturing
We
do not own or operate manufacturing facilities for the production of our current product candidates. We currently rely on third-party
contract manufacturers for all of our required raw materials, manufacturing devices and active pharmaceutical ingredients and for our
preclinical research and clinical trials. Although we are able to manufacture finished product in our Groton Connecticut facility for
our clinical trials, we will rely on third parties for the manufacture of our finished product for commercial sale. We do not have long-term
agreements with any of these third parties. We also do not have any current contractual relationship for the manufacture of Phase 3 clinical
trials or commercial supplies. We intend to enter into agreements with third-party contract manufacturers and one or more backup manufacturers
for future production. We are analyzing the feasibility of building manufacturing capabilities for future development and commercial
quantities of any products that we develop. Such products will need to be manufactured in facilities, and by processes, that comply with
the requirements of the FDA and the regulatory agencies of other jurisdictions in which we are seeking approval.
Competition
The
biopharmaceutical industry is characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary
drugs. While we believe that our knowledge, experience and scientific resources provide us with competitive advantages, we face potential
competition from many different sources, including other biopharmaceutical companies, academic institutions and governmental agencies
as well as public and private research institutions. Any drug candidates that we successfully develop and commercialize will compete
with existing treatments and new treatments that may become available in the future.
Netherton
syndrome
With
respect to Netherton syndrome, no drug has been approved by the FDA, specifically for Netherton syndrome, to date. Standard of care includes
cleansing of the skin with a gentle/soft non-detergent liquid cleansing oil, preferably with an acidic pH (5). Because Netherton syndrome
patient skin is most often dry, scaly and peeling, emollients and moisturizers are also often used. Keratolytics such as salicylic acid,
urea or alpha-hydroxy acids are often irritative and not well tolerated by Netherton syndrome patients. The skin of Netherton syndrome
patients is prone to frequent bacterial infections. Limited infections are treated with topical antibiotics for a short period of up
to 2 weeks. Oral antibiotics may also be used to treat the pathogenic Staphylococcus aureus and Streptococcus strains that
can drive more extreme infections. Bleach baths are also recommended two to three times a week for their antimicrobial effects. Topical
corticosteroids are often used to treat the inflammatory and hyperproliferation associated with non-infected Netherton syndrome lesions,
but due to their adverse effects, must be limited. These adverse events include aminoaciduria, Cushing syndrome, skin atrophy, adrenal
insufficiency, growth retardation, hypertension and weakness. Overuse of topical steroids can even aggravate the defective skin barrier
by inducing loss of the stratum corneum. Systemic retinoids have shown varying degrees of efficacy in Netherton syndrome, but also carry
bone toxicity and teratogenicity as adverse effects. Topical calcineurin inhibitors have been used to reduce erythema (redness) but patients
have shown a tachyphylaxis and reduced efficacy with prolonged treatment. These immunomodulators also carry risks of serious adverse
effects including increased risk of infections, swelling, burning sensations and tingling. Phototherapy (narrowband UVB (NB-UVB) and
psoralen-UVA (PUVA)) has also been investigated in Netherton syndrome patients but has been limited due to its potential to cause erythema
and increases in the risk of skin cancer.
We
are also aware that Sixera Pharma initiated a clinical trial in Europe with SXR-1096, a topical small molecule KLK inhibitor in December
2021 for Netherton syndrome. In addition, Quoin Pharmaceuticals initiated two clinical trials with QRX003, a topical small molecule broad-spectrum
serine protease inhibitor, in December 2022 and March 2023. Krystal Biotech, MatriSys, and BridgeBio have reported they are developing
Netherton syndrome programs that are at a preclinical stage.
Our
commercial opportunity could be reduced or eliminated if our competitors develop and commercialize drugs that are safer, more effective,
have fewer or less severe side effects, are more convenient or are less expensive than ATR-12 or any other drug that we may develop.
Our competitors also may obtain FDA or other regulatory approval for their drugs more rapidly than we may obtain approval for our drug,
which could result in our competitors establishing a strong market position before we are able to enter the market. Many of the companies
against which we are competing, or against which we may compete in the future, have significantly greater financial resources and expertise
in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing
approved drugs than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources
being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors,
particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting
and retaining qualified scientific and management personnel and establishing clinical trial sites and subject registration for clinical
trials, as well as in acquiring technologies complementary to, or that may be necessary for, our programs.
EGFRi-Associated
Rash
To
date, no drug has been specifically approved by the FDA for the treatment of EGFRi-associated rash. The majority of patients (estimated
to be up to 90%) treated continuously with anti-EGFR therapies suffer from dermatological adverse events, especially papulopustular rash,
pruritus (itching), xerosis (dryness), and paronychia (nail infections). Papulopustular or acneiform rash is the most common adverse
event of EGFRis on the skin. This rash negatively impacts compliance with EGFRi treatment in many patients. Dose modification or discontinuation
treatment occurs in severe cases. Because evidence-based controlled trials are still very sparse, treatment of EGFRi skin toxicity primarily
relies on physician experience, and recommendations from expert consensus conferences. As a result, there are geographical variations
and even inconsistencies in the clinical treatment of EGFRi skin rash. Topical corticosteroids are avoided in Europe with respect to
acneiform rash but are often used in the United States. Furthermore, topical treatment is frequently customized to the individual patient
and may change on a case-by-case basis. No topical treatment scheme is universally applicable for all patients.
We
are aware of the following Phase 2 programs developing investigational drug candidates for EGFRi associated rash. Lutris Pharma is developing
LUT014, a topical B-Raf inhibitor, in the US and Israel. Daewoong Pharmaceutical Co. Ltd. is developing DWP708 in Korea.
Intellectual
Property
Overview
We
actively seek to protect our proprietary technology, inventions, improvements to inventions and other intellectual property that is commercially
important to the development of our business by a variety of means, such as seeking, maintaining and defending patent rights, whether
developed internally or licensed from third parties. We also may rely on trade secrets and know-how relating to our proprietary technology
platform, on continuing technological innovation and on future in-licensing opportunities to develop, strengthen and maintain the strength
of our position in the field of gene therapy that may be important for the development of our business. Additional regulatory protection
may also be afforded through data exclusivity, market exclusivity and patent-term extensions where available.
As
of the date of this prospectus, we own or exclusively license three issued U.S. patents, 12 pending U.S. patent applications, three pending PCT application and 57 other foreign patents and patent applications that are important
to the development of our business.
Our
policy is to file patent applications to protect proprietary technology, inventions and improvements to inventions and other intellectual
property that may be commercially important to the development of our business. We also intend to seek additional patent protection or
rely upon know-how or trade secret rights to protect other technologies that may be used to manufacture and develop our live biotherapeutic
products. As described below, we are a party to exclusive license agreements that grant us rights to use specific technologies in our
live biotherapeutic products and in the manufacturing and development of our products.
Our
Patent Portfolio
Our
patent portfolio has broad coverage for therapeutic bacteria pharmaceutical compositions containing these therapeutic bacteria for treating
abnormal skin conditions and methods of making and using these recombinant bacteria. In our broadest filing, we have secured a US patent
that protects pharmaceutical compositions for treating abnormal skin conditions using a bacterial strain expressing a therapeutically
effective amount of a recombinant polypeptide. This patent expires in May 2035. Specifically, this issued patent covers a pharmaceutical
composition containing one or more of the following bacterial strains: Bifidobacterium, Brevibacterium, Propionibacterium, Lactococcus,
Streptococcus, Staphylococcus, Lactobacillus, Enterococcus, Pediococcus, Leuconostoc, or Oenococcus, wherein the bacterial strain has
been engineered to produce a therapeutical polypeptide for treating the abnormal skin conditions. We believe that this patent gives us
broad protection for using recombinant bacteria to treat skin diseases and disorders. through its expiration in May 2035.
Patent
applications directed to our most advanced programs are summarized below.
ATR-12
Our
ATR-12 product candidate is subject to three issued US patents, seven pending US patent applications, and 31 pending foreign
patents and patent applications. These patents and patent applications represent ten families of claims covering, among other aspects,
the pharmaceutical composition of S. epidermidis expressing a recombinant therapeutic polypeptide, the auxotrophic strain of S.
epidermidis, and the recombinant S. epidermidis strain expressing a therapeutic LEKTI protein, and formulations of ATR-12.
One of the issued US patents covers a recombinant bacterial strain containing a therapeutic polypeptide for treating abnormal skin conditions
and expires in 2035. The second issued US patent covers an auxotrophic S. epidermidis that will expire in 2039. If additional
patents were to grant from the pending patent applications, they would expire between 2035 and 2044.
ATR-04
Our
ATR-12 product candidate is subject to one issued US patent, two pending US patent applications, and 17 pending foreign applications.
These patents and patent applications represent two families of claims directed to auxotrophic strains of bacteria and their therapeutic
use for treating disease. We have one issued US patent that covers ATR-04. If additional patents were to grant, they would also expire
in 2039.
Patent
Term and Term Extension
Individual
patents have terms for varying periods depending on the date of filing of the patent application or the date of patent issuance and the
legal term of patents in the countries in which they are obtained. Generally, utility patents issued for applications filed in the United
States are granted a term of 20 years from the earliest effective filing date of a non-provisional patent application. In addition, in
certain instances, the term of a U.S. patent can be extended to recapture a portion of the United States Patent and Trademark Office,
or the USPTO, delay in issuing the patent as well as a portion of the term effectively lost as a result of the FDA regulatory review
period. However, as to the FDA component, the restoration period cannot be longer than five years and the restoration period cannot extend
the patent term beyond 14 years from FDA approval. In addition, only one patent applicable to an approved drug is eligible for the extension,
and only those claims covering the approved drug, a method for using it, or a method of manufacturing may be extended. The duration of
foreign patents varies in accordance with provisions of applicable local law, but typically is also 20 years from the earliest effective
filing date. All taxes, annuities or maintenance fees for a patent, as required by the USPTO and various foreign jurisdictions, must
be timely paid in order for the patent to remain in force during this period of time.
The
actual protection afforded by a patent may vary on a product-by-product basis, from country to country, and can depend upon many factors,
including the type of patent, the scope of its coverage, the availability of regulatory-related extensions and the availability of legal
remedies in a particular country and the validity and enforceability of the patent.
Our
patents and patent applications may be subject to procedural or legal challenges by others. We may be unable to obtain, maintain and
protect the intellectual property rights necessary to conduct our business, and we may be subject to claims that we infringe or otherwise
violate the intellectual property rights of others, which could materially harm our business. For more information, see the section titled
“Risk Factors—Risks Related to Our Intellectual Property.”
Trade
Secrets and Know-How
We
may also rely on trade secrets, know-how, continuing technological innovation and confidential information to develop and maintain our
proprietary position and protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent
protection, including our proprietary processes for manufacturing our live biotherapeutic products. We seek to protect our proprietary
technology and processes, in part, by confidentiality agreements and invention assignment agreements with our employees, consultants,
scientific advisors, contractors and others who may have access to proprietary information, under which they are bound to assign to us
inventions made during the term of their employment or term of service. These agreements may be breached, and we may not have adequate
remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To
the extent that our contractors, commercial partners, collaborators, employees, and consultants use intellectual property owned by others
in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions. For more information, see
the section titled “Risk Factors—Risks Related to Our Intellectual Property.”
We
also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises
and physical and electronic security of our information technology systems.
Exclusive
License Agreement with Fred Hutchison Cancer Center
In
January 2022, we entered into an Exclusive License Agreement with the Fred Hutchinson Cancer Center, or Fred Hutch. Pursuant to our agreement
with Fred Hutch, we obtained an exclusive worldwide license under certain patents related to SyMPL technologies developed and owned by
Fred Hutch to develop, make, manufacture, have manufactured, distribute, have distributed, use, research, improve, import, offer to sell
and sell and otherwise commercialize products that are covered by such patents. Such exclusive license is subject to certain rights retained
by Fred Hutch and the U.S. government. The patent rights licensed to us by Fred Hutch consist of two families of patent applications
directed to methods of bypassing restriction modification systems in order to more easily introduce xenogeneic DNA into engineered microbes.
These patents applications and any patents that issue from these applications will allow us to produce more modified microbes for the
treatment of disease. Our current product candidates do not incorporate the SyMPL technology platform, but we expect that some or all
of our future product candidates will do so. If issued, these two families will expire in 2037 and 2040, respectively.
In
consideration of the license granted to us under the Fred Hutch license agreement, we paid Fred Hutch a nominal upfront payment. In addition,
we are required to pay Fred Hutch certain development and commercial milestone payments and running single digit royalty on net sales
of the licensed products. The Fred Hutch agreement also requires us to reimburse Fred Hutch for the cost of the prosecution and maintenance
of the licensed patents.
Pursuant
to the Fred Hutch license agreement, we are required to use commercially reasonable efforts to bring a licensed product to market through
a vigorous and diligent program for exploitation of the licensed patent rights. The term of the Fred Hutch license agreement will continue
until the later of (i) the expiration of the licensed patents or (ii) ten years from the first sale of a licensed product. We may terminate
the Fred Hutch license agreement at will at any time upon prior written notice to Fred Hutch. Fred Hutch has the right to terminate the
Fred Hutch license agreement if we materially breach the agreement and fail to cure such breach within a specified cure period or if
we become bankrupt or insolvent. For more information related to the intellectual property acquired pursuant to the Fred Hutch license
agreement, see the section titled “Business-Licenses and Intellectual Property Rights.”
We
also hold registered trademarks for our corporate name and design in the U.S. and in seven foreign countries.
Government
Regulations
Pharmaceutical
companies are subject to extensive regulation by foreign, federal, state and local agencies, such as the U.S. FDA, and various similar
agencies in most countries worldwide. The research, development, testing, manufacture, distribution, packaging, labeling, storage, recordkeeping,
marketing and sale of pharmaceutical products are subject to government regulation in the U.S. and various foreign countries. Additionally,
in the U.S., we must follow rules and regulations established by the FDA requiring the presentation of data indicating that our product
candidates are safe and effective and are manufactured in accordance with cGMP regulations. If we do not comply with applicable requirements,
we may be fined, the government may refuse to approve our marketing applications or allow us to manufacture or market our product candidates,
and we may be criminally prosecuted. We, our manufacturers and clinical research organizations may also be subject to regulations under
other foreign, federal, state and local laws, including, but not limited to, the U.S. Occupational Safety and Health Act, the Resource
Conservation and Recovery Act, the Clean Air Act and import, export and customs regulations as well as the laws and regulations of other
countries. The U.S. government has increased its enforcement activity regarding illegal marketing practices domestically and internationally.
As a result, pharmaceutical companies must ensure their compliance with the Foreign Corrupt Practices Act and federal healthcare fraud
and abuse laws, including the False Claims Act.
These
regulatory requirements impact our operations and differ from one country to another, so that securing the applicable regulatory approvals
of one country does not imply the approval of another country. The approval procedures involve high costs and are manpower intensive,
usually extend over many years and require highly skilled and professional resources.
FDA
Market Approval Process
In
the U.S., our product candidates are regulated by the FDA as biologics under the Federal Food, Drug, and Cosmetic Act, or FDCA, the Public
Health Service Act, or PHSA, and regulations promulgated by the FDA. The failure to comply with the applicable requirements at any time
during the product development process, including preclinical testing, clinical testing, the approval process or post-approval process,
may subject an applicant to delays in the conduct of clinical trials, regulatory review and approval, and/or administrative or judicial
sanctions. These sanctions may include, but are not limited to, the FDA’s refusal to allow an applicant to proceed with clinical
testing, refusal to approve pending applications, license suspension or revocation, withdrawal of an approval, warning letters, adverse
publicity, customer notification, product recalls, product seizures, refusal to grant export or import approval, total or partial suspension
of production or distribution, consent decrees, injunctions, fines, and civil or criminal investigations and penalties brought by the
FDA or the U.S. Department of Justice, or other governmental entities.
The
steps usually required to be taken before a new biologic may be marketed in the U.S. generally include:
| |
● |
completion
of preclinical laboratory tests and animal studies performed in accordance with the FDA’s current Good Laboratory Practices
regulation; |
| |
|
|
| |
● |
submission
to the FDA of an IND, which must become effective before clinical trials may begin and must be updated annually or when significant
changes are made; |
| |
|
|
| |
● |
approval
by an independent Institutional Review Board, or IRB, or ethics committee at each treatment site before the trial is commenced; |
| |
|
|
| |
● |
performance
of adequate and well controlled human clinical trials to establish the safety, purity and potency of the proposed biologic product
candidate for its proposed indication for use; |
| |
|
|
| |
● |
submission
of data supporting safety and efficacy as well as detailed information on the manufacture and composition of the product in clinical
development and proposed labeling; |
| |
|
|
| |
● |
preparation
of and submission to the FDA of a BLA after completion of all pivotal clinical trials; |
| |
|
|
| |
● |
satisfactory
completion of an FDA Advisory Committee review, if applicable; |
| |
|
|
| |
● |
a
determination by the FDA within 60 days of its receipt of a BLA to file the application for review; |
| |
|
|
| |
● |
satisfactory
completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the proposed product is produced
to assess compliance with cGMP standards and to assure that the facilities, methods and controls are adequate to preserve the biological
product’s continued safety, purity and potency, and of selected clinical investigation sites to assess compliance with Good
Clinical Practices, or GCP; |
| |
|
|
| |
● |
satisfactory
completion of any FDA audits of the non-clinical and clinical trial sites to assure compliance with CGP requirements and the integrity
of clinical data in support of the BLA; |
| |
|
|
| |
● |
payment
of user fees and securing FDA approval of the BLA for the proposed indication for use; |
| |
|
|
| |
● |
FDA
review and approval of the BLA to permit commercial marketing of the product for particular indications for use in the United States;
and |
| |
● |
compliance
with any post-approval requirements, including REMS and any post-approval studies required by the FDA. |
Preclinical
Studies and Investigational New Drug Application
Preclinical
tests include laboratory evaluations of product chemistry, formulation and stability, as well as animal studies to evaluate the potential
for efficacy and toxicity in animals. The conduct of the preclinical tests and formulation of the compounds for testing must comply with
federal regulations and requirements. The results of the preclinical tests, together with manufacturing information and analytical data,
are submitted to the FDA as part of an IND application. Some preclinical tests may continue even after submission of the IND application.
The IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions
about the product or conduct of the proposed clinical trial, including concerns that human research volunteers will be exposed to unreasonable
health risks. In that case, the IND sponsor and the FDA must resolve any outstanding FDA concerns before the clinical trial can begin.
As
a result, submission of the IND may result in the FDA not allowing the clinical trials to commence or allowing the clinical trial to
commence on the terms originally specified by the sponsor in the IND. If the FDA raises concerns or questions either during this initial
30-day period, or at any time during the IND process, it may choose to impose a partial or complete clinical hold. This order issued
by the FDA would delay either a proposed clinical trial or cause delay in initiation of a phase of an ongoing clinical trial, until all
outstanding concerns have been adequately addressed and the FDA has notified the company that investigations may proceed. This could
cause significant delays or difficulties in completing planned clinical trials in a timely manner.
Clinical
Trials
Clinical
trials involve the administration of the investigational product candidate to healthy volunteers or patients with the disease to be treated
under the supervision of a qualified principal investigator in accordance with good clinical practice, or GCP, requirements. Clinical
studies are conducted under protocols detailing, among other things, the objectives of the study, what types of patients may enter the
study, schedules of tests and procedures, drugs, dosages, and length of study, as well as the parameters to be used in monitoring safety,
and the efficacy criteria to be evaluated. A protocol for each clinical study and any subsequent protocol amendments must be submitted
to the FDA as part of the IND process.
A
sponsor who wishes to conduct a clinical trial outside the U.S. may, but need not, obtain FDA authorization to conduct the clinical trial
under an IND. If a clinical trial outside the U.S. is not conducted under an IND, the sponsor may submit data from the clinical trial
to the FDA in support of a BLA so long as the clinical trial is conducted consistent with the spirit of GCP and in compliance with an
international guideline for the ethical conduct of clinical research known as the Declaration of Helsinki and/or the laws and regulations
of the country or countries in which the clinical trial is performed, whichever provides the greater protection to the participants in
the clinical trial.
An
IRB, either centrally or individually, must also review each clinical trial at each institution at which the clinical trial will be conducted.
The IRB will consider, among other things, clinical trial design, patient informed consent, ethical factors, the safety of human subjects,
the possible liability of the institution, and, where appropriate, the protection of privacy of the human subjects. An IRB must operate
in compliance with the FDA regulations. The FDA, IRB, or the clinical trial sponsor, or the principal investigator may suspend or discontinue
a clinical trial at any time for various reasons, including a finding that the clinical trial is not being conducted in accordance with
FDA requirements or the subjects or patients are being exposed to an unacceptable health risk. Clinical testing also must satisfy extensive
GCP rules and the requirements for informed consent. Additionally, some clinical studies are overseen by an independent group of qualified
experts organized by the clinical study sponsor, known as a data safety monitoring board or committee. This group recommends whether
or not a trial may move forward at designated check points based on access to certain data from the study. The clinical study sponsor
may also suspend or terminate a clinical trial based on evolving business objectives and/or competitive climate.
Clinical
trials are usually conducted in three sequential phases, but the phases may overlap or be combined. Annual progress detailing the results
of the clinical trial phases must be submitted to the FDA.
| |
● |
Phase
1 clinical trials are normally conducted in small groups of healthy volunteers to assess safety and tolerability of various dosing
regimens and pharmacokinetics. For some products for orphan, severe or life-threatening diseases, especially if the product may be
too toxic to administer to healthy humans, the initial clinical trials may be conducted in individuals having a specific disease
for which use the tested product is indicated. These trials in patients are often referred to as Phase 1b trials. If they include
a design to establish a particular dose, they are commonly referred to as Phase 1b/2a clinical trials. Nevertheless, additional Phase
2 (sometimes called Phase 2b) clinical trials are often necessary to refine the final dose chosen to take into a pivotal Phase 3
clinical trial. |
| |
|
|
| |
● |
Phase
2 clinical trials are generally conducted in a limited patient population to identify possible adverse effects and safety risks,
evaluate the efficacy of the product candidate for specific targeted indications and determine does tolerance and optimal dosage.
Multiple Phase 2 clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more costly
Phase 3 clinical trials. |
| |
|
|
| |
● |
Phase
3 clinical trials proceed if the Phase 2 clinical trials demonstrate that a dose range of the product candidate is potentially
effective and has an acceptable safety profile. Phase 3 clinical trials are undertaken to further evaluate, in a larger number of
patients, dosage, provide substantial evidence of clinical efficacy and further test for safety in an expanded and diverse patient
population at multiple, geographically dispersed clinical trial sites. A well-controlled, statistically robust Phase 3 trial may
be designed to deliver the data that regulatory authorities will use to decide whether or not to approve, and, if approved, how to
appropriately label a drug: such Phase 3 studies are referred to as “pivotal.” |
The
FDA may order the temporary or permanent discontinuation of a clinical study at any time or impose other sanctions if it believes that
the clinical study is not being conducted in accordance with FDA requirements or that the participants are being exposed to an unacceptable
health risk. In some cases, the FDA may approve a BLA for a product candidate but require the sponsor to conduct additional clinical
trials to further assess the drug’s safety and effectiveness after BLA approval. Such post-approval trials are typically referred
to as Phase 4 clinical trials. These studies are used to gain additional experience data from the treatment of patients in the intended
therapeutic indication and to document a clinical benefit in the case of drugs or biologics approved under accelerated approval regulations.
If the FDA approves a product while a company has ongoing clinical trials that were not necessary for approval, a company may be able
to use the data from these clinical trials to meet all or part of any Phase 4 clinical trial requirement or to request a change in the
product labeling. Failure to exhibit due diligence with regard to conducting Phase 4 clinical trials could result in withdrawal of approval
for products.
Compliance
with cGMP Requirements
As
a product candidate moves through the clinical testing phases, manufacturing processes are further defined, refined, controlled and validated.
The level of control and validation required by the FDA increases as clinical studies progress. The FDA typically will not approve a
BLA application unless it determines that the manufacturing processes and facilities are in full compliance with cGMP requirements and
able to assure consistent production of the product within required specifications. The PHSA emphasizes the importance of manufacturing
control for products like biologics whose attributes cannot be precisely defined.
We
and the third-party manufacturers on which we rely for the manufacture of our product candidates and their respective components (including
the active pharmaceutical ingredient, or API) are subject to requirements that drugs be manufactured, packaged and labeled in conformity
with cGMPs. To comply with cGMP requirements, manufacturers must continue to spend time, money and effort to meet requirements relating
to personnel, facilities, equipment, production and process, labeling and packaging, quality control, recordkeeping and other requirements.
Manufacturers
and others involved in the manufacture and distribution of products must also register their establishments with the FDA and certain
state regulatory bodies. Both U.S. and non-U.S. manufacturing establishments must register and provide additional information to the
FDA upon their initial participation in the manufacturing process. Any product manufactured by or imported from a facility that has not
registered, whether U.S. or non-U.S., is deemed misbranded under the FDCA. Establishments may be subject to periodic unannounced inspections
by government authorities to ensure compliance with cGMPs and other laws. Inspections must follow a “risk-based schedule”
that may result in certain establishments being inspected more frequently. Manufacturers may also have to provide, on request, electronic
or physical records regarding their establishments. Delaying, denying, limiting, or refusing inspection by the FDA may lead to a product
being deemed to be adulterated.
BLA
Submission and Review
Assuming
completion of all required testing in accordance with all applicable regulatory requirements, detailed information on the product candidate
is submitted to the FDA in the form of a BLA, requesting approval to market the product for one or more indications, together with payment
of a user fee, unless waived. A BLA includes all relevant data available from pertinent nonclinical and clinical studies, including negative
or ambiguous results as well as positive findings, together with detailed information on the chemistry, manufacture, controls and proposed
labeling, among other things. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish
the safety and efficacy of the product candidate for its intended use to the satisfaction of the FDA. The FDA also conducts a pre-approval
inspection of the manufacturer and laboratory prior to approval of the BLA.
If
a BLA submission is accepted for filing, the FDA begins an in-depth review of the BLA. Under the Prescription Drug User Fee Act, or PDUFA,
the FDA’s goal is to complete its initial review and respond to the applicant within ten months of submission, unless the application
relates to an unmet medical need, or is for a serious or life-threatening indication, in which case the goal may be within six months
of BLA submission. However, PDUFA goal dates are not legal mandates, and the FDA response often occurs several months beyond the original
PDUFA goal date. Further, the review process and the target response date under PDUFA may be extended if the FDA requests or the BLA
sponsor otherwise provides additional information or clarification regarding information already provided in the BLA. The BLA review
process can, accordingly, be very lengthy. During its review of a BLA, the FDA may refer the application to an advisory committee for
review, evaluation and recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of
an advisory committee, but it typically follows such recommendations. Data from clinical studies are not always conclusive and the FDA
and/or any advisory committee it appoints may interpret data differently than the applicant.
After
the FDA evaluates the BLA and inspects manufacturing facilities where the drug product and/or its API will be produced and tested, it
will either approve commercial marketing of the drug product with prescribing information for specific indications or issue a complete
response letter indicating that the application is not ready for approval and stating the conditions that must be met in order to secure
approval of the BLA. If the complete response letter requires additional data and the applicant subsequently submits that data, the FDA
nevertheless may ultimately decide that the BLA does not satisfy its criteria for approval. The FDA could also approve the BLA with a
Risk Evaluation and Mitigation Strategies, or REMS, plan to mitigate risks, which could include medication guides, physician communication
plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools.
The FDA also may condition approval on, among other things, changes to proposed labeling, development of adequate controls and specifications,
or a commitment to conduct post-marketing testing. Such post-marketing testing may include Phase 4 clinical trials and surveillance to
further assess and monitor the product’s safety and efficacy after approval. Regulatory approval of products for serious or life-threatening
indications may require that participants in clinical studies be followed for long periods to determine the overall survival benefit
of the drug.
If
the FDA approves one of our product candidates, we will be required to comply with a number of post-approval regulatory requirements.
We would be required to report, among other things, certain adverse reactions and production problems to the FDA, provide updated safety
and efficacy information and comply with requirements concerning advertising and promotional labeling for any of our product candidates.
Also, quality control and manufacturing procedures must continue to conform to cGMPs after approval, and the FDA periodically inspects
manufacturing facilities to assess compliance with cGMPs, which imposes extensive procedural, substantive and record keeping requirements.
If we seek to make certain changes to an approved product, such as certain manufacturing changes, we may need FDA review and approval
before the change can be implemented.
While
physicians may use products for indications that have not been approved by the FDA, we may not label or promote the product for an indication
that has not been approved. Securing FDA approval for new indications is similar to the process for approval of the original indication
and requires, among other things, submitting data from adequate and well-controlled studies that demonstrate the product’s safety
and efficacy in the new indication. Even if such studies are conducted, the FDA may not approve any change in a timely fashion, or at
all.
The
FDA may also require post-marketing testing, or Phase 4 testing, as well as risk minimization action plans and surveillance to monitor
the effects of an approved product or place conditions or an approval that could otherwise restrict the distribution or use of the product.
Fast
Track, Breakthrough Therapy and Priority Review Designations
The
FDA is authorized to designate certain products for expedited review if they are intended to address an unmet medical need in the treatment
of a serious or life-threatening disease or condition, or in the event of an emergency. These programs are fast track designation, breakthrough
therapy designation and priority review designation.
Specifically,
the FDA may designate a product for fast track review if it is intended, whether alone or in combination with one or more other products,
for the treatment of a serious or life-threatening disease or condition, and it demonstrates the potential to address unmet medical needs
for such a disease or condition. For fast track products, sponsors may have greater interactions with the FDA and the FDA may initiate
review of sections of a fast track product’s application before the application is complete. This rolling review may be available
if the FDA determines, after preliminary evaluation of clinical data submitted by the sponsor, that a fast track product may be effective.
The sponsor must also provide, and the FDA must approve, a schedule for the submission of the remaining information and the sponsor must
pay applicable user fees. However, the FDA’s time period goal for reviewing a fast track application does not begin until the last
section of the application is submitted. In addition, the fast track designation may be withdrawn by the FDA if the FDA believes that
the designation is no longer supported by data emerging in the clinical trial process.
Additionally,
in 2012, Congress enacted the Food and Drug Administration Safety and Innovation Act, or FDASIA. This law established a new regulatory
scheme allowing for expedited review of products designated as “breakthrough therapies.” A product may be designated as a
breakthrough therapy if it is intended, either alone or in combination with one or more other products, to treat a serious or life-threatening
disease or condition and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over existing
therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development.
The FDA may take certain actions with respect to breakthrough therapies, including holding meetings with the sponsor throughout the development
process; providing timely advice to the product sponsor regarding development and approval; involving more senior staff in the review
process; assigning a cross-disciplinary project lead for the review team; and taking other steps to design the clinical trials in an
efficient manner.
Moreover,
the FDA may designate a product for priority review if it is a product that treats a serious condition and, if approved, would provide
a significant improvement in safety or effectiveness. The FDA determines, on a case-by-case basis, whether the proposed product represents
a significant improvement when compared with other available therapies. Significant improvement may be illustrated by evidence of increased
effectiveness in the treatment of a condition, elimination or substantial reduction of a treatment-limiting product reaction, documented
enhancement of patient compliance that may lead to improvement in serious outcomes, and evidence of safety and effectiveness in a new
subpopulation. A priority designation is intended to direct overall attention and resources to the evaluation of such applications, and
to shorten the FDA’s goal for taking action on a marketing application from ten months to six months.
Furthermore,
the Secretary of Health and Human Services may authorize unapproved drugs and biologics to be marketed in the event an actual or potential
emergency has been designated by the U.S. government. After an emergency has been designated, the FDA may issue an Emergency Use Authorization,
or EUA, for the use of a specific product based on criteria established by the FDCA. An EUA is product specific and is subject to specific
conditions and restrictions. Once the emergency underlying the EUA ends, then the EUA terminates.
Pediatric
Rare Disease Designation and Priority Review Vouchers
Under
the FDCA, as amended, the FDA incentivizes the development of drugs and biologics that meet the definition of a “rare pediatric
disease,” defined to mean a serious or life-threatening disease in which the serious of life-threatening manifestations primarily
affect individuals aged from birth to 18 years and the disease affects fewer than 200,000 individuals in the United States or affects
200,000 or more in the United States and for which there is no reasonable expectation that the cost of developing and making in the United
States a drug or biologic for such disease or condition will be received from sales in the United States of such drug or biologic. The
sponsor of a product candidate for a rare pediatric disease may be eligible for a voucher that can be used to obtain a priority review
for a subsequent human drug or biologic application after the date of approval of the rare pediatric disease drug product, referred to
as a priority review voucher, or PRV. A sponsor may request rare pediatric disease designation from the FDA prior to the submission of
its NDA or BLA. A rare pediatric disease designation does not guarantee that a sponsor will receive a PRV upon approval of its NDA or
BLA. Moreover, a sponsor who chooses not to submit a rare pediatric disease designation request may nonetheless receive a PRV upon approval
of their marketing application if they request such a voucher in their original. marketing application and meet all of the eligibility
criteria. If a PRV is received, it may be sold or transferred an unlimited number of times. Congress has extended the PRV program until
September 30, 2024, with the potential for PRVs to be granted until 2026.
Post-Approval
Regulation
Once
regulatory approval for marketing of a product or new indication for an existing product is obtained, the sponsor will be required to
comply with post-approval regulatory requirements, including any post-approval requirements that the FDA may have imposed as a condition
of approval. The sponsor will be required to report certain adverse reactions and production problems to the FDA, provide updated safety
and efficacy information and comply with requirements concerning advertising and promotional labeling requirements. Manufacturers and
certain of their subcontractors are required to register their establishments with the FDA and certain state agencies and are subject
to periodic unannounced inspections by the FDA and certain state agencies for compliance with ongoing regulatory requirements, including
cGMP regulations, which impose certain procedural and documentation requirements upon drug manufacturers. Accordingly, the sponsor and
its third-party manufacturers must continue to expend time, money and effort in the areas of production and quality control to maintain
compliance with cGMP regulations and other regulatory requirements.
A
product may also be subject to official lot release, meaning that the manufacturer is required to perform certain tests on each lot of
the product before it is released for distribution. If the product is subject to official release, the manufacturer must submit samples
of each lot, together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the
manufacturer’s tests performed on the lot, to the FDA. The FDA may in addition perform certain confirmatory tests on lots of some
products before releasing the lots for distribution. Finally, the FDA will conduct laboratory research related to the safety, purity,
potency, and effectiveness of pharmaceutical products.
After
an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained
or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse
events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may
result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess
new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among
other things:
| |
● |
restrictions
on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| |
|
|
| |
● |
fines,
warning letters or holds on post-approval clinical trials; |
| |
● |
refusal
of the FDA to approve pending BLAs or supplements to approved BLAs, or suspension or revocation of product license approvals; |
| |
|
|
| |
● |
product
seizure or detention, or refusal to permit the import or export of products; or |
| |
|
|
| |
● |
injunctions
or the imposition of civil or criminal penalties. |
The
FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs and biologics
may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies
actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly
promoted off-label uses may be subject to significant liability.
Orphan
Drug Designation
Under
the Orphan Drug Act, the FDA may grant orphan designation to a drug intended to treat a rare disease or condition affecting fewer than
200,000 individuals in the United States, or in other limited cases. Orphan drug designation (ODD) provides for seven years of market
exclusivity, independent of patent protection, to the company with ODD that brings a particular product to market. In addition, companies
developing orphan drugs are eligible for certain incentives, including tax credits for qualified clinical testing. In addition, a BLA
for a product that has received orphan drug designation is not subject to a prescription drug user fee unless the application includes
an indication other than the rare disease or condition for which the drug was designated.
To
gain exclusivity, if a product that has orphan drug designation subsequently receives the first FDA approval for the disease or condition
for which it has such designation, the product is entitled to the orphan drug exclusivity, which means that the FDA may not approve any
other applications to market the same active moiety for the same indication for seven years, except in limited circumstances, such as
another drug’s showing of clinical superiority over the drug with orphan exclusivity. In addition, doctors may prescribe products
for off-label uses and undermine our exclusivity. Orphan drug exclusivity could block the approval of one of our product candidates for
seven years if a competitor obtains approval for the same active moiety for the same indication before we do, unless we are able to demonstrate
that our product is clinically superior.
A
sponsor may request orphan drug designation of a previously unapproved product or new orphan indication for an already marketed product.
In addition, a sponsor of a product that is otherwise the same product as an already approved orphan drug may seek and obtain orphan
drug designation for the subsequent product for the same rare disease or condition if it can present a plausible hypothesis that its
product may be clinically superior to the first, approved product. More than one sponsor may receive orphan drug designation for the
same product for the same rare disease or condition, but each sponsor seeking orphan drug designation must file a complete request for
designation, and only the first sponsor that obtains approval for that drug for the orphan indication will obtain market exclusivity,
effectively preventing the FDA from approving products under development by competitors for the same drug and same indication, unless
the competitor is able to demonstrate that the product under development is clinically superior to the approved product or the approved
product is not available in sufficient quantities. To permit the FDA to end another manufacturer’s orphan exclusivity period, the
FDA must determine that the manufacturer has demonstrated clinical superiority by showing the later drug is safer, more effective, or
otherwise makes a major contribution to patient care.
The
period of exclusivity begins on the date that the marketing application is approved by the FDA and applies only to the indication for
which the product has been designated. The FDA may approve a second application for the same product for a different use or a subsequent
application for a different drug for the same indication. Orphan drug designation neither shortens the development time or regulatory
review time of a drug nor gives the drug any advantage in the regulatory review or approval process.
We
may plan to pursue orphan drug designation and exclusivity for some of our product candidates in the United States, European Union, and
other geographies of interest for specific products. We cannot guarantee that we will obtain orphan drug designation for any products
in any jurisdiction. Even if we are able to obtain orphan drug designation for a product, we cannot be sure that such product will be
approved, that we will be able to obtain orphan drug exclusivity upon approval, if ever, or that we will be able to maintain any exclusivity
that is granted.
Biosimilars
and Exclusivity
The
Patient Protection and Affordable Care Act, or ACA, which was signed into law on March 23, 2010, included a subtitle called the Biologics
Price Competition and Innovation Act of 2009, or BPCIA. The BPCIA established a regulatory scheme authorizing the FDA to approve biosimilars
and interchangeable biosimilars. The FDA has issued several draft guidance documents outlining an approach to review and approval of
biosimilars. Complexities associated with the larger, and often more complex, structures of biological products, as well as the processes
by which such products are manufactured, pose significant hurdles to implementation, which are still being worked out by the FDA.
Under
the BPCIA, a manufacturer may submit an application for licensure of a biologic product that is “biosimilar to” or “interchangeable
with” a previously approved biological product or “reference product.” In order for the FDA to approve a biosimilar
product, it must find that there are no clinically meaningful differences between the reference product and proposed biosimilar product
in terms of safety, purity, and potency. For the FDA to approve a biosimilar product as interchangeable with a reference product, the
agency must find that the biosimilar product can be expected to produce the same clinical results as the reference product, and (for
products administered multiple times) that the biologic and the reference biologic may be switched after one has been previously administered
without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic.
Under
the BPCIA, an application for a biosimilar product may not be submitted to the FDA until 4 years following the date of approval of the
reference product. The FDA may not approve a biosimilar product until 12 years from the date on which the reference product was approved.
Even if a product is considered to be a reference product eligible for exclusivity, another company could market a competing version
of that product if the FDA approves a full BLA for such product containing the sponsor’s own preclinical data and data from adequate
and well-controlled clinical trials to demonstrate the safety, purity and potency of their product. The BPCIA also created certain exclusivity
periods for biosimilars approved as interchangeable products.
Regulation
Outside the United States
In
order to market any product outside of the United States, a company must also comply with numerous and varying regulatory requirements
of other countries and jurisdictions regarding quality, safety and efficacy that govern, among other things, clinical trials, marketing
authorization, commercial sales and distribution of drug products. Whether or not it obtains FDA approval for a product, the company
would need to obtain the necessary approvals by the comparable non-U.S. regulatory authorities before it can commence clinical trials
or marketing of the product in those countries or jurisdictions. The approval process ultimately varies between countries and jurisdictions
and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other
countries and jurisdictions might differ from and be longer than that required to obtain FDA approval. Regulatory approval in one country
or jurisdiction does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country
or jurisdiction may negatively impact the regulatory process in others.
Regulation
and Marketing Authorization in the European Union
The
European Medicines Agency, or EMA, is the scientific agency of the European Union, or EU, that coordinates the evaluation and monitoring
of new and approved medicinal products such as drugs and biologics. It is responsible for the scientific evaluation of applications for
EU marketing authorizations, as well as the development of technical guidance and the provision of scientific advice to sponsors.
The
process regarding approval of medicinal products in the EU follows roughly the same lines as in the United States and likewise generally
involves satisfactorily completing each of the following:
| |
● |
preclinical
laboratory tests, animal studies and formulation studies all performed in accordance with the applicable EU Good Laboratory Practice
regulations; |
| |
|
|
| |
● |
submission
to the relevant regulatory agencies in EU member states, or national authorities, of a clinical trial application, or CTA, for each
clinical trial, which must be approved before human clinical trials may begin; |
| |
|
|
| |
● |
performance
of adequate and well-controlled clinical trials to establish the safety and efficacy of the product for each proposed indication; |
| |
|
|
| |
● |
submission
to the relevant national authorities of a Marketing Authorisation Application, or MAA, which includes the data supporting safety
and efficacy as well as detailed information on the manufacture and composition of the product in clinical development and proposed
labeling; |
| |
|
|
| |
● |
satisfactory
completion of an inspection by the relevant national authorities of the manufacturing facility or facilities, including those of
third parties, at which the product is produced to assess compliance with cGMP; |
| |
|
|
| |
● |
potential
audits of the non-clinical and clinical trial sites that generated the data in support of the MAA; and |
| |
|
|
| |
● |
review
and approval by the relevant national authority of the MAA before any commercial marketing, sale or shipment of the product. |
Preclinical
Studies
Preclinical
tests include laboratory evaluations of product chemistry, formulation and stability, as well as studies to evaluate the potential efficacy
and toxicity in animals. The conduct of the preclinical tests and formulation of the compounds for testing must comply with the relevant
EU regulations and requirements. The results of the preclinical tests, together with relevant manufacturing information and analytical
data, are submitted as part of the CTA when seeking approval to start a clinical trial, and with the MAA when seeking marketing authorization.
Clinical
Trial Approval
Requirements
for the conduct of clinical trials in the EU including cGCP, are implemented in the currently Clinical Trials Directive 2001/20/EC and
the GCP Directive 2005/28/EC. Pursuant to Directive 2001/20/EC and Directive 2005/28/EC, as amended, a system for the approval of clinical
trials in the EU has been implemented through national legislation of the EU member states. Under this system, approval must be obtained
from the competent national authority in which a trial is planned to be conducted, or in multiple member states if the clinical trial
is to be conducted in a number of member states. To this end, a CTA is submitted, which must be supported by an investigational medicinal
product dossier, or IMPD, and further supporting information prescribed by Directive 2001/20/EC and Directive 2005/28/EC and other applicable
guidance documents. Furthermore, a clinical trial may only be started after a competent ethics committee has issued a favorable opinion
on the clinical trial application in that country.
On
January 31, 2022, the Clinical Trials Regulation (EU) No. 536/2014 replaced the current Clinical Trials Directive 2001/20/EC. To ensure
that the rules for clinical trials are identical throughout the EU, the Clinical Trials Regulation (EU) No. 536/2014 was passed as a
regulation which is directly applicable in all EU member states. The Clinical Trials Directive 2001/20/EC will, however, still apply
three years from the date of entry into application of the Clinical Trials Regulation to (i) clinical trials applications submitted before
the entry into application and (ii) clinical trials applications submitted within one year after the entry into application if the sponsor
opts for the old system.
Regulation
(EU) No 536/2014 aims to simplify and streamline the approval of clinical trial in the EU. The main characteristics of the regulation
include:
| |
● |
A
streamlined application procedure via a single entry point, known as the Clinical Trials Information System; |
| |
|
|
| |
● |
A
single set of documents to be prepared and submitted for the application as well as simplified reporting procedures which will spare
sponsors from submitting broadly identical information separately to various and different national authorities; |
| |
|
|
| |
● |
A
harmonized procedure for the assessment of applications for clinical trials, which is divided in two parts; |
| |
|
|
| |
● |
Strictly
defined deadlines for the assessment of clinical trial application; and |
| |
|
|
| |
● |
The
involvement of the ethics committees in the assessment procedure in accordance with the national law of the member state concerned
but within the overall timelines defined by the Regulation (EU) No 536/2014. |
Marketing
Authorization
Authorization
to market a product in the member states of the EU proceeds under one of four procedures: a centralized procedure, a mutual recognition
procedure, a decentralized procedure or a national procedure.
Centralized
Procedure
The
centralized procedure enables applicants to obtain a marketing authorization that is valid in all EU member states based on a single
application. Certain medicinal products, including products developed by means of biotechnological processes must undergo the centralized
authorization procedure for marketing authorization, which, if granted by the European Commission, based on the opinion of the EMA, is
automatically valid in all EU member states. Sponsors may elect to file an MAA through the centralized procedures for other classes of
products.
The
centralized procedure is mandatory for certain types of products such as, medicines derived from biotechnology processes such as genetic
engineering, advanced-therapy medicines such as gene-therapy or tissue engineered medicine, orphan medicines, and medicinal products
containing a new active substance indicated for the treatment of HIV, AIDS, cancer, diabetes, neurodegenerative disorders, autoimmune
and other immune dysfunctions, and viral diseases.
The
centralized authorization procedure is optional for other medicinal products if they contain a new active substance, if the applicant
shows that the medicinal product concerned constitutes a significant therapeutic, scientific or technical innovation, or that the granting
of authorization is in the public interest of the EU.
Administration
Procedure
Under
the centralized procedure, the EMA’s Committee for Human Medicinal Products, or CHMP serves as the scientific committee that renders
opinions about the safety, efficacy and quality of medicinal products for human use on behalf of the EMA. The CHMP is composed of experts
nominated by each member state’s national authority for medicinal products, with one of them appointed to act as Rapporteur for
the co-ordination of the evaluation with the possible assistance of a further member of the Committee acting as a Co-Rapporteur. After
approval, the Rapporteur(s) continue to monitor the product throughout its life cycle. The CHMP has 210 active days to adopt an opinion
as to whether a marketing authorization should be granted. The process usually takes longer in case additional information is requested,
which triggers clock-stops in the procedural timelines. The process is complex and involves extensive consultation with the regulatory
authorities of member states and a number of experts. When an application is submitted for a marketing authorization in respect of a
drug which is of major interest from the point of view of public health and in particular from the viewpoint of therapeutic innovation,
the applicant may, pursuant to Article 14(9) Regulation (EC) No 726/2004, request an accelerated assessment procedure. If the CHMP accepts
such request, the time-limit of 210 days will be reduced to 150 days, but it is possible that the CHMP can revert to the standard time-limit
for the centralized procedure if it considers that it is no longer appropriate to conduct an accelerated assessment. Once the procedure
is completed, a European Public Assessment Report, or EPAR, is produced. If the opinion is negative, information is given as to the grounds
on which this conclusion was reached. After the adoption of the CHMP opinion, a decision on the MAA must be adopted by the European Commission,
after consulting the EU member states, which in total can take more than 60 days. After a drug has been authorized and launched, it is
a condition of maintaining the marketing authorization that all aspects relating to its quality, safety and efficacy must be kept under
review.
Conditional
Approval
In
specific circumstances, EU legislation (Article 14(7) Regulation (EC) No. 726/2004 and Regulation (EC) No. 507/2006 on Conditional Marketing
Authorizations for Medicinal Products for Human Use) enables applicants to obtain a conditional marketing authorization prior to obtaining
the comprehensive clinical data required for an application for a full marketing authorization. Such conditional approvals may be granted
for products (including medicines designated as orphan medicinal products), if (1) the risk-benefit balance of the product is
positive, (2) it is likely that the applicant will be in a position to provide the required comprehensive clinical trial data, (3) the
product fulfills unmet medical needs, and (4) the benefit to public health of the immediate availability on the market of the medicinal
product concerned outweighs the risk inherent in the fact that additional data are still required. A conditional marketing authorization
may contain specific obligations to be fulfilled by the marketing authorization holder, including obligations with respect to the completion
of ongoing or new studies, and with respect to the collection of pharmacovigilance data. Conditional marketing authorizations are valid
for one year, and may be renewed annually, if the risk-benefit balance remains positive, and after an assessment of the need for additional
or modified conditions and/or specific obligations. The timelines for the centralized procedure described above also apply with respect
to the review by the CHMP of applications for a conditional marketing authorization.
Marketing
Authorization Under Exceptional Circumstances
As
per Article 14(8) Regulation (EC) No 726/2004, products for which the applicant can demonstrate that comprehensive data (in line with
the requirements laid down in Annex I of Directive 2001/83/EC, as amended) cannot be provided (due to specific reasons foreseen in the
legislation) might be eligible for marketing authorization under exceptional circumstances. This type of authorization is reviewed annually
to reassess the risk-benefit balance. The fulfillment of any specific procedures/obligations imposed as part of the marketing authorization
under exceptional circumstances is aimed at the provision of information on the safe and effective use of the product and will normally
not lead to the completion of a full dossier/approval.
Pediatric
Studies
Prior
to obtaining a marketing authorization in the EU, applicants have to demonstrate compliance with all measures included in an EMA-approved
Pediatric Investigation Plan, or PIP, covering all subsets of the pediatric population, unless the EMA has granted (1) a product-specific
waiver, (2) a class waiver, or (3) a deferral for one or more of the measures included in the PIP. The respective requirements for all
marketing authorization procedures are laid down in Regulation (EC) No 1901/2006, the so-called Pediatric Regulation. This requirement
also applies when a company wants to add a new indication, pharmaceutical form or route of administration for a medicine that is already
authorized. The Pediatric Committee of the EMA, or PDCO, may grant deferrals for some medicines, allowing a company to delay development
of the medicine in children until there is enough information to demonstrate its effectiveness and safety in adults. The PDCO may also
grant waivers when development of a medicine in children is not needed or is not appropriate, such as for diseases that only affect the
elderly population.
Before
a marketing authorization application can be filed, or an existing marketing authorization can be amended, the EMA determines that companies
actually comply with the agreed studies and measures listed in each relevant PIP.
Period
of Authorization and Renewals
A
marketing authorization will be valid for five years in principle, and the marketing authorization may be renewed after five years on
the basis of a re-evaluation of the risk-benefit balance by the EMA or by a national authority. To this end, the marketing authorization
holder must provide the EMA or the competent authority with a consolidated version of the file in respect of quality, safety and efficacy,
including all variations introduced since the marketing authorization was granted, at least nine months before the marketing authorization
ceases to be valid. Once renewed, the marketing authorization will be valid for an unlimited period, unless the European Commission or
the national authority decides on justified grounds relating to pharmacovigilance, to proceed with one additional five-year renewal.
Any authorization that is not followed by the actual placing of the drug on the EU market (in case of centralized procedure) or on the
market of the authorizing member state within three years after authorization will cease to be valid, the so-called “sunset clause.”
Orphan
Drug Designation and Exclusivity
The
European Commission can grant orphan medicinal product designation to products for which the sponsor can establish that it is intended
for the diagnosis, prevention, or treatment of (1) a life-threatening or chronically debilitating condition affecting not more
than five in 10,000 people in the EU, or (2) a life threatening, seriously debilitating or serious and chronic condition in the EU and
that without incentives it is unlikely that sales of the drug in the EU would generate a sufficient return to justify the necessary investment.
In addition, the sponsor must establish that there is no other satisfactory method approved in the EU of diagnosing, preventing or treating
the condition, or if such a method exists, the proposed orphan drug will be of significant benefit to patients.
Orphan
drug designation provides a number of benefits, including fee reductions, regulatory assistance, and the possibility to apply for a centralized
EU marketing authorization (see “—Government Regulation and Product Approval—Regulation Outside the United States—Centralized
Authorization Procedure”), as well as 10 years of market exclusivity following a marketing authorization. During this market exclusivity
period, neither the EMA, nor the European Commission nor the Member States can accept an application or grant a marketing authorization
for a medicinal product containing a similar active substance or substances as contained in an authorized orphan medicinal product, and
which is intended for the same therapeutic indication. The market exclusivity period for the authorized therapeutic indication may be
reduced to six years if, at the end of the fifth year, it is established that the orphan drug designation criteria are no longer met,
including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity. In addition,
a competing similar medicinal product may be authorized prior to the expiration of the market exclusivity period, including if it is
shown to be safer, more effective or otherwise clinically superior to the already approved orphan drug or if the holder of the marketing
authorization for the already approved orphan drug is unable to supply sufficient quantities of the product.
If
the MAA of a medicinal product designated as an orphan drug includes the results of all studies conducted in compliance with an agreed
PIP, and a corresponding statement is subsequently included in the marketing authorization granted, the ten-year period of market exclusivity
will be extended to twelve years.
Regulatory
Data Protection
EU
legislation also provides for a system of regulatory data and market exclusivity. Upon receiving marketing authorization, new chemical
entities approved on the basis of complete independent data package benefit from eight years of data exclusivity and an additional two
years of market exclusivity. Data exclusivity prevents regulatory authorities in the EU from referencing the innovator’s data to
assess a generic or biosimilar (abbreviated) application. During the additional two-year period of market exclusivity, a generic or biosimilar
marketing authorization can be submitted, and the innovator’s data may be referenced, but no generic or biosimilar medicinal product
can be marketed until the expiration of the market exclusivity. The overall ten-year period will be extended to a maximum of eleven years
if, during the first eight years of those 10 years, the marketing authorization holder, or MAH, obtains an authorization for one or more
new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical
benefit in comparison with existing therapies. Even if a compound is considered to be a new chemical entity and the innovator is able
to gain the period of data exclusivity, another company nevertheless could also market another version of the drug if such company obtained
marketing authorization based on an MAA with a complete independent data package of pharmaceutical test, preclinical tests and clinical
trials. However, products designated as orphan medicinal products enjoy, upon receiving marketing authorization, a period of 10 years
of orphan market exclusivity (see also “Item 4.B—Government Regulation and Product Approval—Regulation and Marketing
Authorization in the European Union—Orphan Drug Designation and Exclusivity”). Depending upon the timing and duration of
the EU marketing authorization process, products may be eligible for up to five years’ supplementary protection certificates, or
SPCs. Such SPCs extend the rights under the basic patent for the drug.
Regulatory
Requirements After a Marketing Authorization Has Been Obtained
If
we obtain authorization for a medicinal product in the EU, we will be required to comply with a range of requirements applicable to the
manufacturing, marketing, promotion and sale of medicinal products:
Pharmacovigilance
We
will, for example, have to comply with the EU’s stringent pharmacovigilance or safety reporting rules, pursuant to which post-authorization
studies and additional monitoring obligations can be imposed.
Other
requirements relate to, for example, the manufacturing of products and APIs in accordance with good manufacturing practice standards.
EU regulators may conduct inspections to verify our compliance with applicable requirements, and we will have to continue to expend time,
money and effort to remain compliant. Non-compliance with EU requirements regarding safety monitoring or pharmacovigilance, and with
requirements related to the development of products for the pediatric population, can also result in significant financial penalties
in the EU. Similarly, failure to comply with the EU’s requirements regarding the protection of individual personal data can also
lead to significant penalties and sanctions. Individual EU member states may also impose various sanctions and penalties in case we do
not comply with locally applicable requirements.
Manufacturing
The
manufacturing of authorized drugs, for which a separate manufacturer’s license is mandatory, must be conducted in compliance with
the EMA’s cGMP requirements and comparable requirements of other national authorities, which mandate the methods, facilities and
controls used in manufacturing, processing and packing of drugs to assure their safety and identity. The EMA enforces its cGMP requirements
through mandatory registration of facilities and inspections of those facilities. The EMA may have a coordinating role for these inspections
while the responsibility for carrying them out rests with the member states competent authority under whose responsibility the manufacturer
falls. Failure to comply with these requirements could interrupt supply and result in delays, unanticipated costs and lost revenues,
and could subject the applicant to potential legal or regulatory action, including but not limited to warning letters, suspension of
manufacturing, seizure of product, injunctive action or possible civil and criminal penalties.
Marketing
and Promotion
The
marketing and promotion of authorized drugs, including industry-sponsored continuing medical education and advertising directed toward
the prescribers of drugs and/or the general public, are strictly regulated in the EU. The applicable regulations aim to ensure that information
provided by holders of marketing authorizations regarding their products is truthful, balanced and accurately reflects the safety and
efficacy claims authorized by the EMA or by the national authority of the authorizing member state. Failure to comply with these requirements
can result in adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties.
Other
U.S. Healthcare Laws and Compliance Requirements
For
products distributed in the United States, we will also be subject to additional healthcare regulation and enforcement by the federal
government and the states in which we conduct our business. Applicable federal and state healthcare laws and regulations include the
following:
| |
● |
The
federal healthcare anti-kickback statute prohibits, among other things, persons from knowingly and willfully soliciting, offering,
receiving, or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual
for, or the purchase, order, or recommendation of, any good or service, for which payment may be made under federal healthcare programs
such as Medicare, Medicaid, or other governmental programs. A person or entity does not need to have actual knowledge of the federal
anti-kickback statute or specific intent to violate it to have committed a violation; in addition, items or services resulting from
a violation of the federal anti-kickback statute may constitute a false or fraudulent claim for purposes of the False Claims Act; |
| |
● |
The
Ethics in Patient Referrals Act, commonly referred to as the Stark Law, and its corresponding regulations, prohibit physicians from
referring patients for designated health services (including outpatient drugs) reimbursed under the Medicare or Medicaid programs
to entities with which the physicians or their immediate family members have a financial relationship or an ownership interest, subject
to narrow regulatory exceptions, and prohibits those entities from submitting claims to Medicare or Medicaid for payment of items
or services provided to a referred beneficiary; |
| |
|
|
| |
● |
The
federal False Claims Act imposes criminal and civil penalties, including civil whistleblower or qui tam actions, against individuals
or entities for knowingly presenting, or causing to be presented, to the federal government claims for payment that are false or
fraudulent or making a false statement to avoid, decrease, or conceal an obligation to pay money to the federal government; |
| |
|
|
| |
● |
Health
Insurance Portability and Accountability Act of 1996, imposes criminal and civil liability for executing a scheme to defraud any
healthcare benefit program and also imposes obligations, including mandatory contractual terms, with respect to safeguarding the
privacy, security and transmission of individually identifiable health information. This statute also prohibits knowingly and willfully
falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of
or payment for healthcare benefits, items, or services; |
| |
|
|
| |
● |
The
federal false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making
any materially false statement in connection with the delivery of or payment for health care benefits, items or services; |
| |
|
|
| |
● |
The
Physician Payments Sunshine Act, created under the ACA, and its implementing regulations, which requires specified manufacturers
of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s
Health Insurance Program, with specific exceptions, to report annually to the Centers for Medicare & Medicaid Services, or CMS,
information related to payments or other “transfers of value” made to physicians. All such reported information is publicly
available; and |
| |
|
|
| |
● |
Analogous
state laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims
involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers, and some state
laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the
relevant compliance guidance promulgated by the federal government. |
Because
of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our
future business activities could be subject to challenge under one or more of such laws. Efforts to ensure that our business arrangements
with third parties will comply with applicable laws and regulations will involve substantial costs. It is possible that governmental
authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving
applicable fraud and abuse or other health care laws and regulations. If our operations are found to be in violation of any of these
laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative
penalties, damages, fines, exclusion from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or
restructuring of our operations. If any of the physicians or other providers or entities with whom we expect to do business are found
to be not in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions
from government funded health care programs.
Reimbursement
Sales
of our product candidates in the United States may depend, in part, on the extent to which the costs of the product candidates may be
covered by third-party payers, such as government health programs, commercial insurance and managed health care organizations. These
third-party payers are increasingly challenging the prices charged for medical products and services. Additionally, the containment of
health care costs has become a priority of federal and state governments, and the prices of drugs have been a focus in this effort. The
United States government, state legislatures and foreign governments have shown significant interest in implementing cost-containment
programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of
price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures,
could further limit our net revenue and results. If these third-party payers do not consider our product candidates to be cost-effective
compared to other available therapies, they may not cover our product candidates after approval as a benefit under their plans or, if
they do, the level of payment may not be sufficient to allow us to sell our product candidates on a profitable basis.
In
order to secure coverage and reimbursement for any product that might be approved for sale, we may need to conduct expensive pharmacoeconomic
studies in order to demonstrate the medical necessity and cost-effectiveness of the product, in addition to the costs required to obtain
FDA, EMA or other comparable regulatory approvals. Our product candidates may not be considered medically necessary or cost-effective.
A payer’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved.
Third-party reimbursement may not be sufficient to enable us to maintain price levels high enough to realize an appropriate return on
our investment in product development.
Pricing
and reimbursement schemes vary widely from country to country. Some countries provide that drug products may be marketed only after a
reimbursement price has been agreed. Some countries may require the completion of additional studies that compare the cost-effectiveness
of a particular product candidate to currently available therapies. The conduct of such studies could be expensive and result in delays
in our commercializing efforts. The EU provides options for its member states to restrict the range of drug products for which their
national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. EU member states
may approve a specific price for a drug product or may instead adopt a system of direct or indirect controls on the profitability of
the company placing the drug product on the market. Other member states allow companies to fix their own prices for drug products but
monitor and control company profits. The downward pressure on health care costs in general, particularly prescription drugs, has become
very intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries,
cross-border imports from low-priced markets exert competitive pressure that may reduce pricing within a country. There can be no assurance
that any country that has price controls or reimbursement limitations for drug products will allow favorable reimbursement and pricing
arrangements for any of our products.
The
marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party
payors fail to provide adequate coverage and reimbursement. In addition, emphasis on managed care in the United States has increased
and we expect will continue to increase the pressure on drug pricing. Coverage policies, third-party reimbursement rates and drug pricing
regulation may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which
we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Healthcare
Reform
In
the United States, there have been and continue to be a number of significant legislative initiatives to contain healthcare costs. The
ACA was enacted in the United States in March 2010 and contains provisions that may reduce the profitability of drug products, including,
for example, increased rebates for drugs subject to the Medicaid Drug Rebate Program, extension of Medicaid rebates to Medicaid managed
care plans, mandatory discounts for certain Medicare Part D beneficiaries and annual fees based on pharmaceutical companies’ share
of sales to federal health care programs.
In
addition, other legislative changes have been proposed and adopted since the ACA was enacted. These changes included aggregate reductions
to Medicare payments to providers of 2% per fiscal year, effective April 1, 2013 and, due to subsequent legislative amendments to the
statute, will stay in effect through 2027, unless additional Congressional action is taken; however, pursuant to the CARES Act, and subsequent
legislation, these reductions are suspended from May 1, 2020 through March 31, 2022 due to the COVID-19 pandemic. In January 2013, the
American Taxpayer Relief Act of 2012 was signed into law, which, among other things, reduced Medicare payments to several providers,
and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These
new laws may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on
customers for our drugs, if approved, and, accordingly, our financial operations.
Moreover,
recently there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their commercial products.
There have been several recent U.S. Congressional inquiries and proposed and enacted federal and state legislation designed to, among
other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, reduce
the cost of drugs under Medicare, and reform government program reimbursement methodologies for drugs. The FDA released a final rule
on September 24, 2020, effective November 30, 2020, providing guidance for states to build and submit importation plans for drugs from
Canada. Further, on November 20, 2020, the U.S. Department of Health and Human Services, or HHS, finalized a regulation removing safe
harbor protection for price reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy
benefit managers, unless the price reduction is required by law. The rule also creates a new safe harbor for price reductions reflected
at the point-of-sale, as well as a safe harbor for certain fixed fee arrangements between pharmacy benefit managers and manufacturers.
At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical
and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access
and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and
bulk purchasing.
Most
recently, on August 16, 2022, the Inflation Reduction Act of 2022, or the IRA, was signed into law. Among other things, the IRA directs
the Department of Health and Human Services, or the HHS to negotiate the price of certain high-expenditure, single-source drugs and biologics
covered under Medicare. The negotiated prices, which will first become effective in 2026, will be capped at a statutory ceiling price
representing a significant discount from average prices to wholesalers and direct purchasers. The law will also, beginning in 2023, penalize
drug manufacturers that increase prices of Medicare Part B and Part D drugs at a rate greater than the rate of inflation. Further, the
IRA eliminates the “donut hole” under the Medicare Part D program beginning in 2025 by significantly lowering the beneficiary
maximum out-of-pocket cost and creating a new manufacturer discount program. The IRA permits the Secretary of the HHS, to implement many
of these provisions through guidance, as opposed to regulation, for the initial years. HHS has and will continue to issue and update
guidance as these programs are implemented, although the IRA may be subject to legal challenges. It is currently unclear how the IRA
will be implemented but is likely to have a significant impact on the pharmaceutical industry In addition, in response to the Biden administration’s
October 2022 executive order, on February 14, 2023, HHS released a report outlining three new models for testing by the Center for Medicare
and Medicaid Innovation which will be evaluated on their ability to lower the cost of drugs, promote accessibility, and improve quality
of care. It is unclear whether the models will be utilized in any health reform measures in the future.
Although
a number of these, and other proposed measures may require authorization through additional legislation to become effective, Congress
has indicated that it will continue to seek new legislative measures to control drug costs.
CMS
issued a final rule, effective on July 9, 2019, that requires direct-to-consumer advertisements of prescription drugs and biological
products, for which payment is available through or under Medicare or Medicaid, to include in the advertisement the Wholesale Acquisition
Cost, or list price, of that drug or biological product if it is equal to or greater than $35 for a monthly supply or usual course of
treatment. Prescription drugs and biological products that are in violation of these requirements will be included on a public list.
Any
adopted health reform measure could reduce the ultimate demand for our products, if approved, or put pressure on our product pricing.
Individual states in the United States have also become increasingly active in passing legislation and implementing regulations designed
to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product
access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries
and bulk purchasing. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures
to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs.
We expect that additional state and federal healthcare reform measures will be adopted in the future.
We
expect that additional state and federal healthcare reform measures, as well as legal changes by foreign governments, will be adopted
in the future, any of which could limit the amounts that governments will pay for healthcare products and services, which could result
in reduced demand for our product candidates or additional pricing pressures.
Employees
As
of the date of this prospectus, we have 10 employees and full-time consultants, including our executive officers, providing management
and financial services, and general administrative responsibilities. We believe that we maintain a satisfactory working relationship
with our employees, and we have not experienced any significant labor disputes or any difficulty in recruiting staff for our operations.
None of our employees are represented by a labor union.
Human
Capital Resources
Our
human capital resources objectives include, as applicable, identifying, recruiting, retaining, incentivizing and integrating our existing
and new employees, advisors and consultants. The principal purposes of our equity and cash incentive plans are to attract, retain and
reward personnel through the granting of stock-based and cash-based compensation awards, in order to increase stockholder value and the
success of our company by motivating such individuals to perform to the best of their abilities and achieve our objectives.
Legal
Proceedings
We
are not a party to any material legal proceedings. From time to time, we may be involved in legal proceedings or subject to claims incident
to the ordinary course of business. Regardless of the outcome, such proceedings or claims can have an adverse impact on us because of
defense and settlement costs, diversion of resources and other factors, and there can be no assurances that favorable outcomes will be
obtained.
Facilities
Our
executive offices are located in approximately 12,030 square feet of leased office and laboratory space at 21 Business Park Drive, Branford,
Connecticut 06405. The lease expires in 2027, subject to our option to extend the lease for two additional five-year terms. We currently
pay $14,035 per month under the lease, which will increase to $14,385 in 2024, plus our pro rata share of certain operating expenses
of the property.
We
also lease approximately 1,093 square feet of additional laboratory space, which is located at 93 Shennecossett Road, Groton, Connecticut
06340. The lease expires in April 2024, subject to our option to extend the lease for an additional one-year term. We pay $7,235 per
month under the lease plus our pro rata share of certain operating expenses of the property.
We
also lease approximately 1,868 square feet of office and laboratory space, which is located at 500 Cartier Boulevard, Laval, Quebec,
Canada. We pay $6,515 per month under the lease. The lease expires in April 2024, subject to our option to extend the lease for an additional
one-year term.
We
believe that our facilities are adequate to meet our current needs and that additional space can be obtained on commercially reasonable
terms as needed.
MANAGEMENT
Executive
Officers and Directors
The
following table sets forth the names, ages and positions of our current executive officers and directors.
| Name |
|
Age |
|
Position |
| Francisco
D. Salva |
|
53 |
|
President,
Chief Executive Officer and Director |
| Norman
Staskey |
|
54 |
|
Chief
Financial Officer |
| Travis
Whitfill |
|
34 |
|
Chief
Operating Officer and Director |
| Andrew
McClary, MD |
|
38 |
|
Independent
Director |
| Barbara
Ryan |
|
64 |
|
Independent
Director |
| John
Schroer |
|
58 |
|
Independent
Director |
Information
about our Executive Officers and Directors
Francisco
D. Salva has served as our president and chief executive officer and a member of our Board since April 2021. Mr. Salva has over
15 years of experience in senior leadership roles in the biotechnology and pharmaceutical industries. Mr. Salva served as president and
chief executive officer of Complexa, Inc., an inflammation and fibrosis focused biopharmaceutical company, from May 2018 to August 2020.
From February 2011 to November 2016, Mr. Salva served as a co-founder and vice president of operations of Acerta Pharma B.V., Inc, a
cancer and autoimmune focused biopharmaceutical company. Mr. Salva serves as a director of Vincerx Pharma, Inc. (Nasdaq: VINC). Prior
to his operating roles, Mr. Salva served in various senior positions in the venture capital and investment banking industries focusing
on healthcare, biotechnology and pharmaceuticals companies. Mr. Salva received a B.A. from Brown University and an MSc in economics and
philosophy from the London School of Economics. We believe that Mr. Salva’s experience as a senior executive, venture capitalist
and investment banker in the biotech and pharmaceutical industries qualifies him to serve on our Board.
Norman
Staskey has served as our chief financial officer since October 2022. Since May 15, 2021, Mr. Staskey has also served as a senior
director of Danforth Advisors, a national consulting firm providing financial, accounting and reporting services to the life science
industry. From September 2014 to May 2021, Mr. Staskey was employed by EY (formally Ernst & Young), most recently as a managing director
in EY’s Financial Accounting and Advisory services practice.
Travis
Whitfill is a co-founder of Azitra and has served on our Board since inception. Mr. Whitfill has served in various roles at Azitra,
including chief scientific officer from January 2014 to September 2019 and director of advanced technology since September 2019, and
as chief operating officer since June 2023. Mr. Whitfill served as a partner at Bios Equity Partners, LP, a biotechnology-focused venture
capital firm, from October 2015 to June 2023 and a senior analyst at Bios Research from September 2014 to June 2023. He has also served
as an associate research scientist and assistant professor adjunct at Yale University from July 2016 to March 2022 and since March 2022,
respectively, with appointments in the Departments of Pediatrics and Emergency Medicine. Mr. Whitfill has served on the board of directors
of IN8Bio, Inc. (Nasdaq: INAB) since March 2018, 410 Medical from September 2017 to July 2019 and SIRPant Immunotherapeutics from September
2021 to June 2023. Mr. Whitfill has led numerous grant-funded projects, holds several patents and has co-authored over 60 publications.
Mr. Whitfill received a B.S. from Dallas Baptist University, an MPH from Yale University and an MPhil from University College London.
We believe that Mr. Whitfill’s strong background in entrepreneurship and in the biotech and healthcare industries qualifies him
to serve on our Board.
Andrew
McClary, MD has served as a member of our Board since March 2019. Dr. McClary is the founding general partner at KdT Ventures
LP, a biotechnology focused venture firm founded in 2017. At KdT, Dr. McClary invests in companies leveraging the intersection of the
physical sciences and engineering, both computational and biochemical. In addition to leading KdT’s investment in Azitra, Dr. McClary
has also led KdT’s investments in PathAI, Dyno Therapeutics, Solugen, Terray Therapeutics, STRM Therapeutics, Elegen, and Checkerspot.
Prior to KdT, Dr. McClary served on faculty as a Pathologist at Stanford University and was an early employee at Included Health (formerly
known as Grand Rounds), where he was the physician lead for the Data Science and Analytics team. Dr. McClary received his M.D. from Tulane
University, where he has held prior academic appointments and was an HHMI/NIH Fellow, and his Sc.B. in Biochemistry and Molecular Biology
from Brown University. We believe that Dr. McClary’s medical and scientific expertise as a physician-scientist coupled with his
experience working in the venture capital industry qualifies him to serve on our Board.
Barbara
Ryan has served as a member of our Board since June 2023. Ms. Ryan founded Barbara Ryan Advisors, a capital markets and communications
firm, in 2012 following a more than 30-year career on Wall Street as a sell-side research analyst covering the U.S. pharmaceutical industry.
Ms. Ryan has deep experience in equity and debt financings, M&A, valuation, SEC reporting, financial analysis and corporate strategy
across a broad range of life sciences companies. Ms. Ryan worked on several of the industry’s largest M&A transactions, including
Shire’s defense versus a hostile takeover attempt by Abbvie, Shire’s takeover of Baxalta, Allergan’s defense against
Valeant and Perrigo’s defense versus Mylan Ms. Ryan served as an executive team member and on the disclosure committee for Radius
Health from January 2014 to December 2017. Previously, Ms. Ryan was a managing director at Deutsche Bank/Alex Brown and head of the company’s
pharmaceutical research team for 19 years and began her research career covering the pharmaceutical industry at Bear Stearns in 1982.
Ms. Ryan currently serves as a director on the board of MiNK Therapeutics, Inc. (Nasdaq: INKT), where she chairs the audit committee,
INVO Bioscience, Inc, (Nasdaq: INVO), Invidior, PLC (LON:INDV) and The Red Door Community (formerly Gilda’s Club NYC), a non-profit
organization. Ms. Ryan is the founder of Fabulous Pharma Females, a non-profit whose mission is to advance women in the biopharma industry,
is a member of the editorial advisory board of Pharmaceutical Executive Magazine, a faculty member of the GLG Institute and a member
of the Prix Galien executive advisory board. We believe that Ms. Ryan is qualified to serve as a member of our Board because of her experience
and knowledge of corporate finance, mergers and acquisitions, corporate governance, as well as other operational, financial and accounting
matters gained as a past and present executive officer and/or director of other public and private companies.
John
Schroer has served as a member of our Board since June 2023. Mr. Schroer has served as chief financial officer of Alumis, Inc.,
a privately held biotechnology company developing precision immunology therapies, since March 2022. Mr. Schroer was chief financial officer
of Arsenal Biosciences, Inc., a privately held biotechnology company developing programmable cell therapy for solid tumors, from February
2021 to February 2022. Mr. Schroer was chief financial officer of Translate Bio, Inc., a biotechnology company developing mRNA therapeutics
and vaccines acquired by Sanofi in September 2021 for $3.2 billion, from May 2018 to December 2020. Previously, Mr. Schroer was Sector
Head – Global Health Care for Allianz Global Investors, an international asset management firm, from January 2014 to May 2018.
Mr. Schroer received his B.S. and M.B.A from the University of Wisconsin – Madison. We believe that Mr. Schroer’s strong
background holding leadership positions in the biotechnology industry and almost 30 years of investing in the life sciences sector qualifies
him to serve on our Board.
Family
Relationships
There
are no family relationships among any of our executive officers or directors.
Involvement
in Certain Legal Proceedings
To
the best of our knowledge, none of our executive officers or directors were involved in any legal proceedings described in Item 401(f)
of Regulation S-K in the past ten years.
Board
Composition
Our
Board may establish the authorized number of directors from time to time by resolution. Our Board consists of five members, three of
whom qualify as independent under the listing rules of the NYSE American. Neither Mr. Salva nor Mr. Whitfill are considered to be independent
due to their roles as executive officers of the Company. However, our Board has determined that Dr. McClary, Ms. Ryan and Mr. Schroer
do not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director
and that he or she is “independent” as that term is defined under the applicable rules and regulations of the SEC and the
listing requirements and rules of the NYSE American. In making this determination, our Board considered the current and prior relationships
that each of Dr. McClary, Ms. Ryan and Mr. Schroer has with our Company and all other facts and circumstances our Board deemed relevant
in determining their independence, including their beneficial ownership of our capital stock.
Role
of the Board in Risk Oversight
One
of the key functions of our Board is informed oversight of our risk management process. Our Board does not have a standing risk management
committee, but rather intends to administer this oversight function directly through the board of directors as a whole, as well as through
various standing committees of our Board that will address risks inherent in their respective areas of oversight. In particular, our
Board will be responsible for monitoring and assessing strategic risk exposure and our audit committee will have the responsibility to
consider and discuss our major financial risk exposures and the steps our management has taken to monitor and control these exposures,
including guidelines and policies to govern the process by which risk assessment and management is undertaken. The audit committee will
also monitor compliance with legal and regulatory requirements.
Board
Committees
Our
Board has established an audit committee, compensation committee and nominating and corporate governance committee, each of which operates
pursuant to a committee charter. Our Board may establish other committees to facilitate the management of our business. The composition
and functions of each committee are described below.
Audit
Committee
Our
audit committee consists of Dr. McClary, Ms. Ryan and Mr. Schroer, with Mr. Schroer serving as chairperson. Our Board has determined
that each member meets the independence requirements of the Sarbanes-Oxley Act, Rule 10A-3 under the Exchange Act and the applicable
listing standards of the NYSE American. Each member of our audit committee can read and understand fundamental financial statements in
accordance with the SEC and NYSE American audit committee requirements. In arriving at this determination, the Board has examined each
audit committee member’s scope of experience and the nature of their prior and/or current employment.
Our
Board has determined that Mr. Schroer qualifies as an audit committee financial expert within the meaning of SEC regulations and meets
the financial sophistication requirements of the NYSE American listing rules. Both our independent registered public accounting firm
and management periodically meet with our audit committee.
The
functions of this committee include, among other things:
| ● | select
a qualified firm to serve as the independent registered public accounting firm to audit our
financial statements; |
| ● | discuss
the scope and results of the audit with the independent registered public accounting firm,
and review, with management and the independent registered public accounting firm, our interim
and year-end operating results; |
| ● | reviewing
and approving the engagement of our independent auditors to perform audit services and any
permissible non-audit services; |
| ● | develop
procedures for employees to submit concerns anonymously about questionable accounting or
audit matters; |
| ● | review
our policies on risk assessment and risk management; |
| ● | review
related-party transactions; and |
| ● | reviewing
and evaluating on an annual basis the performance of the audit committee and the audit committee
charter. |
We
believe that the composition and functioning of our audit committee complies with all applicable requirements of the Sarbanes-Oxley Act,
and all applicable SEC and NYSE American rules and regulations. We intend to comply with future requirements to the extent they become
applicable to us.
Compensation
Committee
Our
compensation committee consists of Ms. Ryan and Mr. Schroer, with Ms. Ryan serving as chairperson. Each of the members is a non-employee
director, as defined in Rule 16b-3 promulgated under the Exchange Act. Our Board has determined that each member is “independent”
as defined under the applicable listing standards of the NYSE American, including the standards specific to members of a compensation
committee. The functions of this committee include, among other things:
| ● | reviewing,
modifying and approving (or if it deems appropriate, making recommendations to the full Board
regarding) our overall compensation strategy and policies; |
| ● | reviewing,
modifying and approving (or if it deems appropriate, making recommendations to the full Board
regarding) the compensation and other terms of employment of our executive officers; |
| ● | reviewing
and approving (or if it deems it appropriate, making recommendations to the full Board regarding)
the equity incentive plans, compensation plans and similar programs advisable for us, as
well as modifying, amending or terminating existing plans and programs; |
| ● | administering
our equity incentive plans; |
| ● | establishing
policies with respect to equity compensation arrangements; and |
| ● | reviewing
and evaluating on an annual basis the performance of the compensation committee and the compensation
committee charter. |
We
believe that the composition and functioning of our compensation committee complies with all applicable requirements of the Sarbanes-Oxley
Act, and all applicable SEC and the NYSE American rules and regulations. We intend to comply with future requirements to the extent they
become applicable to us.
Nominating
and Corporate Governance Committee
Our
nominating and corporate governance committee consists of Dr. McClary and Ms. Ryan, with Dr. McClary serving as chairperson. The composition
of our nominating and corporate governance committee meets the requirements for independence under the NYSE American listing standards
and SEC rules and regulations. The functions of this committee include, among other things:
| ● | identifying,
evaluating and making recommendations to our board of directors regarding nominees for election
to our board of directors and its committees; |
| ● | evaluating
the performance of our board of directors and of individual directors; |
| ● | considering
and making recommendations to our board of directors regarding the composition of our board
of directors and its committees; |
| ● | reviewing
developments in corporate governance practices; |
| ● | evaluating
the adequacy of our corporate governance practices and reporting; and |
| ● | developing
and making recommendations to our board of directors regarding corporate governance guidelines
and matters. |
Each
of our committees operates under a written charter that satisfies the applicable listing requirements and rules of the NYSE American.
Code
of Ethics
We
have adopted a Code of Ethics applicable to all of our employees, executive officers and directors. The Code of Ethics is available on
our website at www.azitrainc.com. The audit committee of our Board is responsible for overseeing the Code of Ethics and must approve
any waivers of the Code of Ethics for employees, executive officers and directors. In addition, we have posted on our website all disclosures
that are required by law or the listing standards of the applicable stock exchange concerning any amendments to, or waivers from, any
provision of the Code of Ethics.
Executive
Compensation
Officer
Compensation
The
following table sets forth the compensation awarded to or earned by our chief executive officer and our two other highest paid executive
officers for the years ended December 31, 2023 and 2022. In reviewing the table, please note that:
| ● | Francisco
D. Salva was appointed to serve as our president and chief executive officer in April 2021; |
| ● | Norman
Staskey was appointed to serve as our chief financial officer in October 2022; and |
| ● | Travis
Whitfill was appointed to serve as our chief operating officer in June 2023. |
| | |
Year | | |
Salary ($) | | |
Bonus ($) | | |
Option Awards $ (1) | | |
All Other Compensation (2) | | |
Total | |
| Francisco D. Salva, | |
| 2023 | | |
$ | 420,000 | | |
$ | — | | |
$ | — | | |
$ | 7,030 | | |
$ | 427,030 | |
| Pres. and CEO | |
| 2022 | | |
$ | 403,846 | | |
$ | — | | |
$ | — | | |
$ | 202 | | |
$ | 404,048 | |
| Norman Staskey, | |
| 2023 | | |
$ | — | | |
$ | — | | |
$ | 20,700 | | |
$ | 314,025 | | |
$ | 334,725 | |
| CFO | |
| 2022 | | |
$ | — | | |
$ | — | | |
$ | — | | |
$ | 63,200 | | |
$ | 63,200 | |
| Travis Whitfill, | |
| 2023 | | |
$ | 158,846 | | |
$ | — | | |
$ | 20,700 | | |
$ | 2,767 | | |
$ | 182,313 | |
| COO | |
| 2022 | | |
$ | -- | | |
$ | — | | |
$ | — | | |
$ | 10,050 | | |
$ | 10,050 | |
| (1) | The
dollar amounts in the Option Awards columns above reflect the values of options as of the
grant date for the years ended December 31, 2023 and 2022, in accordance with
ASC 718, Compensation-Stock Compensation (“ASC 718”) and, therefore, do
not necessarily reflect actual benefits received by the individuals. Assumptions used in
the calculation of these amounts are included in Note 11 to our audited financial statements. |
| (2) | All
other compensation includes commuter benefits, vacation payouts, relocation reimbursements,
401K match contributions, and life insurance premiums, plus consulting fees paid for Mr.
Staskey’s services as chief financial officer and consulting fees paid to Mr. Whitfill
prior to his appointment as chief operating officer. |
Narrative
Disclosure to Officer Compensation Table
All
of our current named executive officers are at-will employees and set forth below is a summary of the current terms of their compensatory
arrangements.
Francisco
D. Salva
We
have entered into an executive employment agreement dated April 22, 2021 with Mr. Salva, pursuant to which Mr. Salva serves as our president
and chief executive officer. We have agreed to pay Mr. Salva an annual base salary of $420,000 under the agreement. Mr. Salva is also
eligible to receive a bonus of up to 35% of his base salary based on performance parameters set by our Board. Mr. Salva’s executive
employment agreement entitles him to participate in health insurance and other benefits, at our expense, made available to other executive
officers. In the event of Mr. Salva’s termination by us without cause or his resignation for good reason, as such terms are defined
in the executive employment agreement, Mr. Salva will be entitled to the continuation of his base salary and health insurance coverage
for a period of 12 months and a prorated amount of his annual bonus for the year in which the termination occurred, subject to the achievement
of applicable performance targets. Mr. Salva’s executive employment agreement is an “at will” agreement subject to
termination by either party at any time and for any reason, subject to certain notice requirements. The agreement contains customary
provisions relating to intellectual property assignment, confidentiality and indemnification.
In
connection with our execution of the executive employment agreement, we granted to Mr. Salva an option to purchase up to 465,760 shares
of our common stock at an exercise price of $1.56 per share under the 2016 Plan. The options vest and become exercisable as follows:
80% of the options, or options to purchase 372,608 shares of our common stock, are subject to time-based vesting, with options to purchase
93,152 shares (25%) vesting on the first anniversary of the grant and options to purchase 279,456 shares (75%) vesting in equal monthly
installments over the 36 months following the first anniversary; 20% of the options, or options to purchase 13,120 shares of our common
stock, shall vest upon patient dosing in the first in-human clinical trial of ATR-12 or a substitute live biotherapeutic product, as
determined by our Board in its reasonable discretion. The options expire on the ten-year anniversary of the date of grant.
Norman
Staskey
Mr.
Staskey serves as our chief financial officer pursuant to a Consulting Agreement dated October 12, 2002 between us and Danforth Advisors,
LLC. Pursuant to the Consulting Agreement, Danforth Advisors provides to us certain strategic and financial advice and support services,
including Mr. Staskey’s services as chief financial officer, at hourly rates between $135 and $575 per hour, depending on the level
of service and the seniority of the service provider. The Consulting Agreement is subject to termination by either party on 30 days written
notice and contains customary provisions relating to intellectual property assignment, confidentiality and indemnification.
On September 8, 2023, we granted
to Mr. Staskey an option to purchase up to 10,000 shares of our common stock at an exercise price of $2.07 per share under the 2016 Plan.
The options are subject to time-based vesting, with options to purchase 2,500 shares (25%) vesting on the first anniversary of the grant
and options to purchase 7,500 shares (75%) vesting in equal monthly installments over the 36 months following the first anniversary;
The options expire on the ten-year anniversary of the date of grant.
Travis
Whitfill
We
have entered into an executive employment agreement dated July 5, 2023 with Mr. Whitfill, pursuant to which Mr. Whitfill serves as our
chief operating officer. We have agreed to pay Mr. Whitfill an annual base salary of $350,000 under the agreement. Mr. Whitfill is also
eligible to receive a bonus of up to 30% of his base salary based on performance parameters set by our Board. Mr. Whitfill’s executive
employment agreement entitles him to participate in health insurance and other benefits, at our expense, made available to other executive
officers. In the event of Mr. Whitfill’s termination by us for any reason other than cause or his incapacity, as such terms are
defined in the executive employment agreement, Mr. Whitfill will be entitled to the continuation of his base salary for a period of six
months and, if unpaid at the time of termination, his annual bonus for the year prior to the year in which the termination occurred.
Mr. Whitfill’s executive employment agreement is an “at will” agreement subject to termination by either party at any
time and for any reason, subject to certain notice requirements. The agreement contains customary provisions relating to intellectual
property assignment, confidentiality and indemnification.
On
September 8, 2023, we granted to Mr. Whitfill an option to purchase up to 10,000 shares of our common stock at an exercise price of $2.07
per share under the 2016 Plan. The options are subject to time-based vesting, with options to purchase 2,500 shares (25%) vesting on
the first anniversary of the grant and options to purchase 7,500 shares (75%) vesting in equal monthly installments over the 36 months
following the first anniversary; The options expire on the ten-year anniversary of the date of grant.
Non-Employee
Director Compensation
On
September 8, 2023, we granted to Barbara Ryan and John Schroer each an option to purchase up to 10,000 shares of our common stock at
an exercise price of $2.07 per share under the 2016 Plan. The options are subject to time-based vesting, with options to purchase 2,500
shares (25%) vesting on the first anniversary of the grant and options to purchase 7,500 shares (75%) vesting in equal monthly installments
over the 36 months following the first anniversary, subject t their continued service on our board of directors; The options expire on
the ten-year anniversary of the date of grant.
Other
than the options described above, we have not paid
any directors’ fees or other compensation to our directors for their services as directors. All of our directors receive reimbursement
for out-of-pocket expenses for attending board of directors meetings. We intend to commence the payment of our non-executive directors,
including the payment of cash and equity awards, or a combination of both, but we have not adopted any such plans or policies as of this
date. From time to time, we may also engage certain outside members of the board of directors to perform services on our behalf and we
will compensate such persons for the services which they perform.
Stock
Incentive Plans
We
have adopted the Azitra, Inc. 2016 Stock Incentive Plan, or 2016 Plan, providing for the grant of non-qualified stock options and incentive
stock options to purchase shares of our common stock and for the grant of restricted and unrestricted share grants and restricted stock
units. We currently have reserved 242,345 shares of our common stock under the 2016 Plan. The purpose of the 2016 Plan is to provide
eligible participants with an opportunity to acquire an ownership interest in our company. All officers, directors, employees and consultants
to our company are eligible to participate under the 2016 Plan. The 2016 Plan provides that options may not be granted at an exercise
price less than the fair market value of our shares of common stock on the date of grant. As of the date of this prospectus, we have
outstanding options granted under the 2016 Plan to purchase an aggregate of 1,248,255 shares of our common stock at an average exercise
price of $1.34 per share.
In
March 2023, our Board and stockholders approved and adopted the Azitra, Inc. 2023 Stock Incentive Plan, or 2023 Plan, providing for the
grant of non-qualified stock options and incentive stock options to purchase shares of our common stock and for the grant of restricted
and unrestricted share grants and restricted stock units. We currently have reserved 2,000,000 shares of our common stock under the 2023
Plan. The purpose of the 2023 Plan is to provide eligible participants with an opportunity to acquire an ownership interest in our company.
All officers, directors, employees and consultants to our company are eligible to participate under the 2023 Plan. The 2023 Plan provides
that options may not be granted at an exercise price less than the fair market value of our shares of common stock on the date of grant.
As of the date of this prospectus, we have outstanding options granted under the 2023 Plan to purchase an aggregate of 40,000 shares
of our common stock at an average exercise price of $2.07 per share.
Related
Party Transactions
Except
as set forth below, since January 1, 2020, we have not been a party to any transaction in which the amount involved in the transaction
exceeded the lesser of $120,000 or one percent of the average of our total assets as of December 31, 2022 and 2021, and in which any
of our directors, executive officers or, to our knowledge, beneficial owners of more than 5% of our voting securities or any member of
the immediate family of any of the foregoing persons had or will have a direct or indirect material interest, other than compensation
arrangements, which include equity and other compensation, termination, change in control and other arrangements, which are described
under “Executive Compensation.” We have adopted a policy that any transactions with directors, officers, beneficial
owners of five percent or more of our common stock, any immediate family members of the foregoing or entities of which any of the foregoing
are also officers or directors or in which they have a financial interest, will only be on terms consistent with industry standards and
approved by a majority of the disinterested directors of our Board.
In
September 2022, we issued unsecured convertible promissory notes in the aggregate principal amount of $4.35 million to five existing
stockholders, including notes in the aggregate principal amount of $4 million to three funds under common control, namely Bios Fund III,
LP, Bios Fund III QP, LP, and Bios Fund III NT, LP. The Bios entities beneficially own a collective 4,883,658 shares, or approximately
43.0%, of our outstanding common stock immediately prior to this offering. In connection the Bios entities investment in our Company,
we granted the Bios entities certain board appointment rights pursuant to which they appointed to our Board a Bios representative who
served on our Board from April 2016 to the date preceding the date of our June 2023 IPO. In addition, Travis Whitfill, our co-founder,
chief operating officer and a member of our Board, was a partner of Bios Equity Partners, LP, the general partner of the aforementioned
Bios entities, until June 2023.
In
December 2019, we entered into a Joint Development Agreement, or JDA, with Bayer pursuant to which we agreed to the joint development
of certain strains selected from our proprietary microbial library. Bayer paid us a one-time low six figure payment upon execution of
the JDA. Pursuant to the JDA, Bayer is responsible for reimbursing us for our development costs, and in 2023 Bayer has paid us a six-figure
dollar amount for our development costs. We have granted Bayer an option to acquire an exclusive royalty bearing license for up to six
(6) strains subject to development activities under the JDA, including an exclusive royalty bearing license to any related patent rights.
In
September 2020, Bayer’s venture capital group, LEAPS by Bayer, purchased $8 million of our Series B preferred stock. In connection
with the investment, we granted Bayer certain board appointment rights pursuant to which they appointed to our Board a Bayer representative
who served on our Board from September 2020 to the date preceding the date of this prospectus.
Indemnification
Agreements
We
have entered into indemnification agreements with each of our directors and executive officers that may be broader than the specific
indemnification provisions contained in the DGCL. These indemnification agreements require us, among other things, to indemnify our directors
and executive officers against liabilities that may arise by reason of their status or service. These indemnification agreements also
require us to advance all expenses incurred by the directors and executive officers in investigating or defending any such action, suit,
or proceeding. We believe that these agreements are necessary to attract and retain qualified individuals to serve as directors and executive
officers.
Limitation
of Liability of Directors and Indemnification of Directors and Officers
The
Delaware General Corporation Law provides that corporations may include a provision in their certificate of incorporation relieving directors
of monetary liability for breach of their fiduciary duty as directors, provided that such provision shall not eliminate or limit the
liability of a director (i) for any breach of the director’s duty of loyalty to the corporation or its stockholders, (ii) for acts
or omissions not in good faith or which involve intentional misconduct or a knowing violation of law, (iii) for unlawful payment of a
dividend or unlawful stock purchase or redemption, or (iv) for any transaction from which the director derived an improper personal benefit.
Our amended and restated certificate of incorporation provides that directors are not liable to us or our stockholders for monetary damages
for breach of their fiduciary duty as directors to the fullest extent permitted by Delaware law. In addition to the foregoing, our amended
and restated certificate of incorporation provides that we shall indemnify directors and officers to the fullest extent permitted by
law.
The
above provisions in our amended and restated certificate of incorporation may have the effect of reducing the likelihood of derivative
litigation against directors and may discourage or deter stockholders or management from bringing a lawsuit against directors for breach
of their fiduciary duty, even though such an action, if successful, might otherwise have benefited us and our stockholders. However,
we believe that the foregoing provisions are necessary to attract and retain qualified persons as directors.
Insofar
as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to our directors, officers and controlling
persons pursuant to the foregoing provisions, or otherwise, we have been advised that in the opinion of the SEC, such indemnification
is against public policy as expressed in the Securities Act and is, therefore, unenforceable.
PRINCIPAL
STOCKHOLDERS
The
following table sets forth certain information regarding the beneficial ownership of our shares of common stock as of the date of this
prospectus by:
| ● | each
person who is known by us to be the beneficial owner of more than five percent (5%) of our
issued and outstanding shares of common stock; |
| ● | each
of our executive officers and directors; and |
| ● | all
of the aforementioned directors and executive officers as a group. |
The
beneficial ownership of each person was calculated based on 12,097,643 shares of common stock issued and outstanding prior to the offering.
The SEC has defined “beneficial ownership” to mean more than ownership in the usual sense. For example, a person has beneficial
ownership of a share not only if he owns it, but also if he has the power (solely or shared) to vote, sell or otherwise dispose of the
share. Beneficial ownership also includes the number of shares that a person has the right to acquire within 60 days of the date of this
prospectus, pursuant to the exercise of options or warrants or the conversion of notes, debentures or other indebtedness. Two or more
persons might count as beneficial owners of the same share. Percentage ownership of our common stock after this offering assumes our
sale of shares of common stock in this offering and no exercise of the underwriters’ over-allotment option.
Unless
otherwise indicated, the address for each reporting person is c/o Azitra, Inc., 21 Business Park Drive, Branford, Connecticut 06405.
| Name of
Director and Executive Officer | |
Number
of Shares | | |
Percentage
Owned Prior to Offering | | |
Percentage
Owned After Offering | |
| Francisco D. Salva
(1) | |
| 268,929 | | |
| 2.2 | % | |
| 1.8 | % |
| Norman Staskey | |
| — | | |
| * | | |
| * | |
| Travis Whitfill (2) | |
| 412,653 | | |
| 3.4 | % | |
| 2.7 | % |
| Andrew McClary (3) | |
| 313,713 | | |
| 2.6 | % | |
| 2.1 | % |
| Barbara Ryan | |
| — | | |
| * | | |
| * | |
| John Schroer | |
| — | | |
| * | | |
| * | |
| Directors and executive
officers, as a group (6 persons) | |
| 995,295 | | |
| 8.0 | % | |
| 6.4 | % |
| Name and
Address of Five Percent Stockholders | |
Number
of Shares | | |
Percentage
Owned Prior to Offering | | |
Percentage
Owned After Offering | |
| Bios Equity Entities
(4) | |
| 6,799,021 | | |
| 55.5 | % | |
| 44.6 | % |
| Bayer Health Care, LLC (5) | |
| 1,307,401 | | |
| 10.8 | % | |
| 8.7 | % |
| Connecticut Innovations, Inc.
(6) | |
| 814,031 | | |
| 6.7 | % | |
| 5.4 | % |
| * |
Represents less than 1% of
the number of shares of our common stock outstanding. |
| (1) | Includes
263,929 shares of our common stock issuable upon exercise of presently exercisable options. |
| | |
| (2) | Includes
93,153 shares issuable upon exercise of presently exercisable options. |
| | |
| (3) | The
securities are held by KdT Ventures LP, of which Mr. McClary is the managing partner. Includes
21,802 shares of our common stock issuable upon the exercise of warrants. |
| | |
| (4) | Consists
of (i) 799,467 shares of common stock held by Bios Fund I, LP, (ii) 436,173 shares of common
stock and 39,760 shares of common stock issuable upon the exercise of warrants held by Bios
Azitra Co-Invest I, LP, (iii) 313,250 shares of common stock and 22,726 shares of common
stock issuable upon the exercise of warrants held by Bios Fund II, LP, (iv) 404,767 shares
of common stock held by Bios Fund III, LP, (v) 467,613 shares of common stock held by Bios
Fund I QP, LP, (vi) 1,023,442 shares of common stock and 74,236 shares of common stock issuable
upon the exercise of warrants held by Bios Fund II QP, LP and 74,236 shares of common stock
issuable upon the exercise of warrants, (vii) 2,643,705 shares of common stock held by Bios
Fund III QP, LP, (viii) 137,000 shares of common stock and 9,938 shares of common stock issuable
upon the exercise of warrants held by Bios Fund II NT, LP, (ix) 426,944 shares of common
stock held by Bios Fund III NT, LP. Bios Equity Partners, LP is the general partner of the
following entities: Bios Fund I, LP and Bios Fund I QP, LP. Bios Equity Partners II, LP is
the general partner of Bios Fund II, LP, QP, LP, Bios Fund II, LP and Bios Fund II NT, LP.
Cavu Management, LP, an entity managed and controlled by Mr. Les Kreis, and Bios Capital
Management, LP, an entity managed and controlled by Mr. Aaron Fletcher, are the general partners
of Bios Equity I, LP and Bios Equity II, LP. Cavu Advisors LLC, an entity that is managed
and controlled by Mr. Kreis, is the general partner of Cavu Management LP. Bios Advisors
GP, LLC, an entity that is managed and controlled by Mr. Fletcher, is the general partner
of Bios Capital Management, LP. The shares owned by Bios Fund I, Bios Fund I QP, Bios Fund
II, Bios Fund II QP, Bios Fund II NT and Bios Fund III NT (“Bios Equity Entities”)
are aggregated for purposes of reporting share ownership information. Mr. Kreis and Mr. Fletcher
share voting and investment control with respect to shares held by the Bios Equity Entities.
Travis Whitfill, a chief operator officer and director of the Company, was a partner at Bios
Equity Partners, LP until June 2023, but did not have any voting or investment control with
respect to the shares held by the Bio Equity Entities. The address for Bios Equity Entities
is 1751 River Run, Suite 400, Fort Worth, Texas 76107. |
| (5) | The
address for Bayer HealthCare, LLC is 610 Main Street Cambridge, Massachusetts 02139. |
| (6) | Includes
41,314 shares of common stock issuable upon the exercise of warrants. The address for Connecticut
Innovations, Inc. is 470 James Street, Suite 8 New Haven, CT 06513. |
DESCRIPTION
OF SECURITIES
General
The
following description summarizes the most important terms of our capital stock. Because it is only a summary, it does not contain all
the information that may be important to you. For a complete description of the matters set forth in this “Description of Securities,”
you should refer to our amended and restated certificate of incorporation and amended and restated bylaws and investor rights agreement,
which are included as exhibits to the registration statement of which this prospectus forms a part, and to the applicable provisions
of Delaware law.
Our
authorized capital stock consists of 100,000,000 shares of common stock, $0.0001 par value per share, and 10,000,000 shares of undesignated
preferred stock, $0.0001 par value per share.
There
are 12,097,643 shares of our common stock outstanding and no shares of our preferred stock outstanding as of the date of this prospectus.
As
of the date of this prospectus, we had 32 stockholders of record.
Common
Stock
The
holders of common stock are entitled to one vote for each share of common stock. The holders of common stock are entitled to any dividends
that may be declared by the Board out of funds legally available for payment of dividends at such times and in such amounts as the Board
in its discretion. In the event of any liquidation, dissolution or winding up of the Company, holders of common stock are entitled to
receive the assets of the Company available for distribution to its stockholders ratably in proportion to the number of shares of common
stock held by the holders of common stock. The holders of shares of common stock have no preemptive, conversion, subscription rights
or cumulative voting rights.
Preferred
Stock
As
of the date of this prospectus, there are a total of 10,000,000 shares of undesignated preferred stock authorized for issuance, none
of which are outstanding.
Our
Board is authorized, without further action by our stockholders, to provide from time to time out of the unissued shares of preferred
stock for one or more series of preferred stock, and with respect to each such series, to fix the number of shares constituting such
series and the designation of such series, the powers (including voting powers), if any, of the shares of such series and the preferences
and relative, participating, optional, special or other rights, if any, and the qualifications, limitations, or restrictions, if any,
of the shares of such series. The issuance of our preferred stock could adversely affect the voting power of holders of our common stock
and the likelihood that such holders will receive dividend payments and payments upon liquidation. In addition, the issuance of preferred
stock could have the effect of delaying, deferring, or preventing a change of control or other corporate action.
Pre-Funded
Warrants
The
following summary of certain terms and provisions of the Pre-Funded Warrants that are being offered hereby is not complete and is subject
to, and qualified in its entirety by, the provisions of the Pre-Funded Warrants, the form of which is filed as an exhibit to the registration
statement of which this prospectus forms a part. Prospective investors should carefully review the terms and provisions of the form of
Pre-Funded Warrants for a complete description of the terms and conditions of the Pre-Funded Warrants.
Duration
and Exercise Price
Each
Pre-Funded Warrant offered hereby will have an initial exercise price equal to $0.001 per share of common stock. The Pre-Funded Warrants
will be exercisable immediately, and may be exercised at any time until the Pre-Funded Warrants are exercised in full. The exercise price
and number of shares of common stock issuable upon exercise is subject to appropriate proportional adjustment in the event of share dividends,
share splits, reorganizations or similar events affecting our shares of common stock and the exercise price.
Exercisability
The
Pre-Funded Warrants will be exercisable, at the option of each holder, in whole or in part by delivering to us a duly executed exercise
notice and within the earlier of (i) two trading days and (ii) the number of trading days comprising the standard settlement period with
respect to the shares of common stock as in effect on the date of delivery of the notice of exercise thereafter, payment in full for
the number of shares of common stock purchased upon such exercise (except in the case of a cashless exercise as discussed below). A holder
may not exercise any portion of the Pre-Funded Warrant to the extent that the holder, together with its affiliates and any other persons
acting as group together with any such persons, would own more than 4.99% (or, at the election of the purchaser, 9.99%) of the number
of shares of our common stock outstanding immediately after exercise (the “Beneficial Ownership Limitation”); provided that
a holder with Beneficial Ownership Limitation of 4.99%, upon notice to use and effective 61 days after the date such notice is delivered
to us may increase the Beneficial Ownership Limitation so long as it in no event exceeds 9.99% of the number of shares of common stock
outstanding immediately after exercise.
Cashless
Exercise
The
Pre-Funded Warrants may also be exercised, in whole or in part, at such time by means of “cashless exercise” in which the
holder shall be entitled to receive upon such exercise (either in whole or in part) the net number of shares of common stock determined
according to a formula set forth in the Pre-Funded Warrants, which generally provides for a number of shares of common stock equal to
(A)(1) the last closing trade price for such security on NYSE American on (x) the trading day immediately preceding the date of the applicable
notice of exercise, if the notice of exercise is executed and delivered on day that is not a trading day or prior to the opening of “regular
trading hours” on a trading day or (y) the trading day of the notice of exercise, if the notice of exercise is executed and delivered
after the close of “regular trading hours” on such trading day; (2) at the option of the holder, either (x) the volume
weighted average price on the trading day immediately preceding the date of the applicable notice of exercise or (y) the bid price of
our common stock on NYSE American as of the time of the holder’s execution of the applicable notice of exercise if such notice
of exercise is executed during “regular trading hours” on a trading day and is delivered within two hours thereafter (including
until two hours after the close of “regular trading hours” on a trading day); or (3) the last closing trade price for such
security on NYSE American on the day of the notice of exercise, if the notice of exercise is executed and delivered after “regular
trading hours” on a trading day, less (B) the exercise price, multiplied by (C) the number of shares of common stock the Pre-Funded
Warrant was exercisable into, with such product then divided by the number determined under clause (A) in the this sentence.
Fractional
Shares
No
fractional shares of common stock will be issued upon the exercise of the Pre-Funded Warrants. Rather, we will, at our election, either
pay a cash adjustment in respect of such final fraction in an amount equal to such fraction multiplied by the exercise price or round
up to the next whole shares of common stock.
Transferability
Subject
to applicable laws, a Pre-Funded Warrant may be transferred at the option of the holder upon surrender of the Pre-Funded Warrant to us
together with the appropriate instruments of transfer and funds sufficient to pay any transfer taxes payable upon such transfer.
Trading
Market
There
is no trading market available for the Pre-Funded Warrants on any securities exchange or nationally recognized trading system. We do
not intend to list the Pre-Funded Warrants on any securities exchange or nationally recognized trading system. The shares of common stock
issuable upon exercise of the Pre-Funded Warrants are currently listed on NYSE American under the symbol “AZTR.”
Rights
as a Shareholder
Except
as otherwise provided in the Pre-Funded Warrants or by virtue of such holder’s ownership of the underlying shares of common stock,
the holders of the Pre-Funded Warrants do not have the rights or privileges of holders of our ordinary shares represented by shares of
common stock, including any voting rights, until they exercise their Pre-Funded Warrants.
Fundamental
Transaction
In
the event of a fundamental transaction, as described in the Pre-Funded Warrants and generally including any reorganization, recapitalization
or reclassification of our shares of common stock, the sale, transfer or other disposition of all or substantially all of our properties
or assets, our consolidation or merger with or into another person, the acquisition of more than 50% of our outstanding shares of common
stock or 50% or more of the voting power of the common equity of the Company, the holders of the Pre-Funded Warrants will be entitled
to receive upon exercise of the Pre-Funded Warrants the kind and amount of securities, cash or other property that the holders would
have received had they exercised the Pre-Funded Warrants immediately prior to such fundamental transaction.
Warrants
We
have outstanding the following warrants to purchase shares of our common stock:
| ● | Warrants
issued in connection with our April 2018 placement of unsecured convertible promissory notes
to purchase up to an aggregate of 47,890 shares of our common stock, at a per share exercise
price equal to $0.48. The warrants expire in April 2028. |
| ● | Warrants
issued in connection with our February 2019 placement of Series A-1 convertible preferred
shares to purchase up to an aggregate of 215,856 shares of our common stock, at a
per share exercise price equal to $5.28. The warrants expire in February 2029. |
| ● | Warrants
issued to the underwriter of our IPO to purchase 60,000 shares of our common stock. This
warrant is exercisable at $6.25 per share. The warrants expire in June 2028. |
Stock
Incentive Plans
We
have adopted the Azitra, Inc. 2016 Stock Incentive Plan, or 2016 Plan, providing for the grant of non-qualified stock options and incentive
stock options to purchase shares of our common stock and for the grant of restricted and unrestricted share grants and restricted stock
units. We currently have reserved 242,345 shares of our common stock under the 2016 Plan. The purpose of the 2016 Plan is to provide
eligible participants with an opportunity to acquire an ownership interest in our company. All officers, directors, employees and consultants
to our company are eligible to participate under the 2016 Plan. The 2016 Plan provides that options may not be granted at an exercise
price less than the fair market value of our shares of common stock on the date of grant. As of the date of this prospectus, we have
outstanding options granted under the 2016 Plan to purchase an aggregate of 1,248,255 shares of our common stock at an average exercise
price of $1.34 per share.
In
March 2023, our Board and stockholders approved and adopted the Azitra, Inc. 2023 Stock Incentive Plan, or 2023 Plan, providing for the
grant of non-qualified stock options and incentive stock options to purchase shares of our common stock and for the grant of restricted
and unrestricted share grants and restricted stock units. We currently have reserved 2,000,000 shares of our common stock under the 2023
Plan. The purpose of the 2023 Plan is to provide eligible participants with an opportunity to acquire an ownership interest in our company.
All officers, directors, employees and consultants to our company are eligible to participate under the 2023 Plan. The 2023 Plan provides
that options may not be granted at an exercise price less than the fair market value of our shares of common stock on the date of grant.
As of the date of this prospectus, we have outstanding options granted under the 2023 Plan to purchase an aggregate of 40,000 shares
of our common stock at an average exercise price of $2.07 per share.
Dividends
We
do not anticipate the payment of cash dividends on our common stock in the foreseeable future.
Registration
Rights
Certain
holders of our common stock, or their permitted transferees, are entitled to the registration rights described below. The registration
of shares of our common stock pursuant to the exercise of registration rights described below would enable the holders to sell these
shares without restriction under the Securities Act when the applicable registration statement is declared effective. We will pay the
registration expenses, other than the underwriting discounts and commissions, of the shares registered pursuant to the registrations
described below. The registration rights described below will expire upon the earlier of June 20, 2026 or when all investors, considered
with their affiliates, can sell all of their shares in a three-month period under Rule 144.
Convertible
Preferred Stock Registration Rights. In connection with our convertible preferred stock financings, we entered into an investor rights
agreement, as amended, pursuant to which we have granted the purchasers of our convertible preferred stock certain demand and piggyback
respiration rights. Those parties beneficially hold approximately 9,542,519 shares of our common stock, including 1,838,775 shares of
our common stock issuable upon exercise of warrants issued to the parties in connection with our 2018 placement of our unsecured convertible
promissory notes and our 2019 placement of Series A-1 convertible preferred shares.
Pursuant
to the investor rights agreement, we are required, upon the written request by the holders of at least 50% of the shares that are entitled
to registration rights under the investor rights agreement, to register, as soon as practicable, all or a portion of these shares for
public resale. We are required to effect two demand registrations pursuant to a registration statement on Form S-1.
Subject to our eligibility to use a registration statement on Form S-3, we are required to effect an unlimited number of demand registrations
pursuant to Form S-3, provided such requests for registration be for an aggregate offering price, net of the underwriting discounts and
commissions, equal or greater than $1 million. Pursuant to the investor rights agreement, we have also granted to the piggyback registration
rights and demand registration rights. These demand and piggyback registration rights terminate as to each investor when their shares
subject to the registration rights agreement may be sold by the investor pursuant to Rule 144 under the Securities Act without regard
to both the volume limitations for sales as provided in Rule 144.
Underwriter
Registration Rights. In connection with our IPO, we issued to the representative of the underwriters or its designees warrants, referred
to as the Representative’s Warrants, to purchase up to a total of 60,000 shares of our common stock. The Representative’s
Warrants provide for registration rights (including a one-time demand registration right and unlimited piggyback rights) consistent with
FINRA Rule 5110.05. The demand for registration may be made at any time beginning on the initial exercise date of the Representative’s
Warrants and expiring on the fifth anniversary of the effective date of this registration statement in accordance with FINRA Rule 5110(g)(8)(C).
In addition to the one-time demand registration right, the Representative’s Warrants has unlimited piggyback rights, for a period
of no more than two years from the initial exercise date of the Representative’s Warrants in accordance with FINRA Rule 5110(g)(8)(D).
Anti-Takeover
Effects of Certain Provisions of Delaware Law and Our Charter Documents
The
following is a summary of certain provisions of Delaware law and our amended and restated certificate of incorporation and amended and
restated bylaws. This summary does not purport to be complete and is qualified in its entirety by reference to the corporate law of Delaware
and our amended and restated certificate of incorporation and amended and restated bylaws.
Delaware
Law
We
are subject to Section 203 of the Delaware General Corporation Law, an anti-takeover law. In general, Section 203 prohibits a Delaware
corporation from engaging in any business combination (as defined below) with any interested stockholder (as defined below) for a period
of three years following the date that the stockholder became an interested stockholder, unless:
| ● | prior
to that date, the board of directors of the corporation approved either the business combination
or the transaction that resulted in the stockholder becoming an interested stockholder; |
| ● | upon
consummation of the transaction that resulted in the stockholder becoming an interested stockholder,
the interested stockholder owned at least 85% of the voting stock of the corporation outstanding
at the time the transaction commenced, excluding for purposes of determining the number of
shares of voting stock outstanding (but not the voting stock owned by the interested stockholder)
those shares owned by persons who are directors and officers and by excluding employee stock
plans in which employee participants do not have the right to determine whether shares held
subject to the plan will be tendered in a tender or exchange offer; or |
| ● | on
or subsequent to that date, the business combination is approved by the board of directors
of the corporation and authorized at an annual or special meeting of stockholders, and not
by written consent, by the affirmative vote of at least 66 2/3% of the outstanding voting
stock that is not owned by the interested stockholder. |
Section
203 defines “business combination” to include the following:
| ● | any
merger or consolidation involving the corporation and the interested stockholder; |
| ● | any
sale, transfer, pledge or other disposition of 10% or more of the assets of the corporation
involving the interested stockholder; |
| ● | subject
to certain exceptions, any transaction that results in the issuance or transfer by the corporation
of any stock of the corporation to the interested stockholder; |
| ● | subject
to limited exceptions, any transaction involving the corporation that has the effect of increasing
the proportionate share of the stock of any class or series of the corporation beneficially
owned by the interested stockholder; or |
| ● | the
receipt by the interested stockholder of the benefit of any loans, advances, guarantees,
pledges or other financial benefits provided by or through the corporation. |
In
general, Section 203 defines an interested stockholder as any entity or person beneficially owning 15% or more of the outstanding voting
stock of the corporation, or who beneficially owns 15% or more of the outstanding voting stock of the corporation at any time within
a three-year period immediately prior to the date of determining whether such person is an interested stockholder, and any entity or
person affiliated with or controlling or controlled by any of these entities or persons.
Our
Charter Documents
Our
charter documents include provisions that could have the effect of discouraging others from making tender offers for our shares and may
have the effect of deterring hostile takeovers or delaying changes in our control or management. These provisions are intended to enhance
the likelihood of continued stability in the composition of our Board and its policies and to discourage certain types of transactions
that may involve an actual or threatened acquisition of us. These provisions are also designed to reduce our vulnerability to an unsolicited
acquisition proposal and to discourage certain tactics that may be used in proxy fights. However, such provisions may have the effect
of discouraging, delaying or preventing a change in control or an unsolicited acquisition proposal that a stockholder might consider
favorable, including a proposal that might result in the payment of a premium over the market price for the shares held by our stockholders.
Certain of these provisions are summarized in the following paragraphs.
Effects
of authorized but unissued common stock and preferred stock. One of the effects of the existence of authorized but unissued common
stock and preferred stock may be to enable our Board to make more difficult or to discourage an attempt to obtain control of our Company
by means of a merger, tender offer, proxy contest or otherwise, and thereby to protect the continuity of management. If, in the due exercise
of its fiduciary obligations, the Board were to determine that a takeover proposal was not in our best interest, such shares could be
issued by the Board without stockholder approval in one or more transactions that might prevent or render more difficult or costly the
completion of the takeover transaction by diluting the voting or other rights of the proposed acquirer or insurgent stockholder group,
by putting a substantial voting block in institutional or other hands that might undertake to support the position of the incumbent Board,
by effecting an acquisition that might complicate or preclude the takeover, or otherwise.
Cumulative
Voting. Our amended and restated certificate of incorporation does not provide for cumulative voting in the election of directors,
which would allow holders of less than a majority of the stock to elect some directors.
Vacancies.
Our amended and restated bylaws provide that all vacancies may be filled by the affirmative vote of a majority of directors then in office,
even if less than a quorum.
Special
Meeting of Stockholders and Stockholder Action by Written Consent. A special meeting of stockholders may only be called by our Board
or the chairperson of our Board. All stockholder actions must be effected at a duly called meeting of stockholders and not by written
consent.
Advance
Notice Provisions. Our amended and restated bylaws provide advance notice procedures for stockholders seeking to bring business before
our annual meeting of stockholders or to nominate candidates for election as directors at our annual meeting of stockholders. Our amended
and restated bylaws will also specify certain requirements regarding the form and content of a stockholder’s notice. These provisions
might preclude our stockholders from bringing matters before our annual meeting of stockholders or from making nominations for directors
at our annual meeting of stockholders if the proper procedures are not followed. We expect that these provisions may also discourage
or deter a potential acquirer from conducting a solicitation of proxies to elect the acquirer’s own slate of directors or otherwise
attempting to obtain control of our company.
Choice
of Forum. Our amended and restated certificate of incorporation and amended and restated bylaws will provide that the Court of Chancery
of the State of Delaware will be the exclusive forum for any derivative action or proceeding brought on our behalf; any action asserting
a breach of fiduciary duty; any action asserting a claim against us arising pursuant to the Delaware General Corporation Law, our amended
and restated certificate of incorporation or our bylaws; or any action asserting a claim against us that is governed by the internal
affairs doctrine.
Transfer
Agent and Registrar
The
transfer agent and registrar for our shares of common stock is VStock Transfer, LLC. The transfer agent and registrar’s address
is 18 Lafayette Place, Woodmere, New York 11598.
National
Securities Exchange Listing
Our
common stock is listed on the NYSE American under the symbol “AZTR.”
SHARES
ELIGIBLE FOR FUTURE SALE
Future
sales of substantial amounts of shares of common stock, including shares issued upon the exercise of outstanding warrants and options,
in the public market after this offering, or the possibility of these sales occurring, could adversely affect the prevailing market price
for our common stock or impair our ability to raise equity capital.
Upon
the completion of this offering, a total of 15,097,643. shares of common stock will be outstanding. All 3,000,000 shares
of common stock sold in this offering by us, plus any shares sold upon exercise of the underwriter’s over-allotment option, will
be freely tradable in the public market without restriction or further registration under the Securities Act, unless these shares are
held by “affiliates,” as that term is defined in Rule 144 under the Securities Act. In addition, of the 12,097,643 shares
of our common stock outstanding prior to this offering, approximately 3,984,145 shares will be freely tradable in the public market without
restriction or further registration under the Securities Act.
The
remaining 8,113,498 shares of common stock will be “restricted securities,” as that term is defined in Rule 144 under the
Securities Act. These restricted securities are eligible for public sale only if they are registered under the Securities Act or if they
qualify for an exemption from registration under Rule 144 under the Securities Act, which is summarized below.
Subject
to the lock-up agreements described below and the provisions of Rule 144 under the Securities Act, these restricted securities are available
for sale in the public market.
Rule
144
In
general, under Rule 144 as currently in effect, a person who is not deemed to have been one of our affiliates for purposes of the Securities
Act at any time during the 90 days preceding a sale and who has beneficially owned the shares proposed to be sold for at least six months,
including the holding period of any prior owner other than our affiliates, is entitled to sell such shares without complying with the
manner of sale, volume limitation, or notice provisions of Rule 144, subject to compliance with the public information requirements of
Rule 144. If such a person has beneficially owned the shares proposed to be sold for at least one year, including the holding period
of any prior owner other than our affiliates, then such person is entitled to sell such shares without complying with any of the requirements
of Rule 144.
In
general, under Rule 144, as currently in effect, our affiliates or persons selling shares on behalf of our affiliates are entitled to
sell upon expiration of the lock-up agreements described below, within any three-month period a number of shares that does not exceed
the greater of:
| ● | 1%
of the number of shares of common stock then outstanding; or |
| ● | the
average weekly trading volume of the common stock during the four calendar weeks preceding
the filing of a notice on Form 144 with respect to such sale. |
Sales
under Rule 144 by our affiliates or persons selling shares on behalf of our affiliates are also subject to certain manner of sale provisions
and notice requirements and to the availability of current public information about us.
Lock-Up
Agreements
Each
of our directors and officers have agreed, subject to certain exceptions, not to offer, pledge, sell, contract to sell, grant, lend,
or otherwise transfer or dispose of, directly or indirectly, or enter into any swap or other arrangement that transfers to another, in
whole or in part, any of the economic consequences of ownership of any shares of our capital stock or any securities convertible into
or exercisable or exchangeable for shares of our common stock, for a period of twelve (12) months after our IPO, or until June 15, 2024,
without the prior written consent of the representative. We, subject to certain exceptions, have agreed, subject to certain exceptions,
not offer, pledge, sell, contract to sell, grant, lend, or otherwise transfer or dispose of, directly or indirectly, or enter into any
swap or other arrangement that transfers to another, in whole or in part, any of the economic consequences of ownership of any shares
of our capital stock or any securities convertible into or exercisable or exchangeable for shares of our common stock, for a period of
ninety (90) days after the date of this prospectus, without the prior written consent of the representative.
Equity
Plans
We
have filed with the SEC a registration statement on Form S-8 under the Securities Act to register all of the shares of common stock to
be issued or reserved for issuance under our 2016 Plan and 2023 Plan. Shares covered by this registration statement are eligible for
sale in the public market, upon the expiration or release from the terms of the lock-up agreements and subject to vesting of such shares.
MATERIAL
U.S. FEDERAL INCOME TAX CONSEQUENCES FOR NON-U.S. HOLDERS
The
following discussion is a summary of the material U.S. federal income tax consequences to Non-U.S. Holders (as defined below) of the
purchase, ownership, and disposition of our common stock issued pursuant to this offering but does not purport to be a complete analysis
of all potential tax effects. The effects of other U.S. federal tax laws, such as estate and gift tax laws, and any applicable state,
local, or non-U.S. tax laws are not discussed. This discussion is based on the U.S. Internal Revenue Code of 1986, as amended, or the
Code, Treasury Regulations promulgated thereunder, judicial decisions, and published rulings and administrative pronouncements of the
U.S. Internal Revenue Service, or the IRS, in each case in effect as of the date hereof. These authorities may change or be subject to
differing interpretations. Any such change or differing interpretation may be applied retroactively in a manner that could adversely
affect a Non-U.S. Holder. We have not sought and will not seek any rulings from the IRS regarding the matters discussed below. There
can be no assurance the IRS or a court will not take a contrary position to that discussed below regarding the tax consequences of the
purchase, ownership, and disposition of our common stock.
This
discussion is limited to Non-U.S. Holders that hold our common stock as a “capital asset” within the meaning of Section 1221
of the Code (generally, property held for investment). This discussion does not address all U.S. federal income tax consequences relevant
to a Non-U.S. Holder’s particular circumstances, including the impact of the Medicare contribution tax on net investment income
and the alternative minimum tax. In addition, it does not address consequences relevant to Non-U.S. Holders subject to special rules,
including, without limitation:
| ● | U.S.
expatriates and former citizens or long-term residents of the United States; |
| ● | persons
holding our common stock as part of a hedge, straddle, or other risk reduction strategy or
as part of a conversion transaction or other integrated investment; |
| ● | banks,
insurance companies, and other financial institutions; |
| ● | brokers,
dealers, or traders in securities; |
| ● | “controlled
foreign corporations,” “passive foreign investment companies,” and corporations
that accumulate earnings to avoid U.S. federal income tax; |
| ● | partnerships
or other entities or arrangements treated as partnerships for U.S. federal income tax purposes
(and investors therein); |
| ● | tax-exempt
organizations or governmental organizations; |
| ● | persons
deemed to sell our common stock under the constructive sale provisions of the Code; |
| ● | persons
who hold or receive our common stock pursuant to the exercise of any employee stock option
or otherwise as compensation; |
| ● | tax-qualified
retirement plans; and |
| ● | “qualified
foreign pension funds” as defined in Section 897(l)(2) of the Code and entities all
of the interests of which are held by qualified foreign pension funds. |
If
an entity treated as a partnership for U.S. federal income tax purposes holds our common stock, the tax treatment of a partner in the
partnership will depend on the status of the partner, the activities of the partnership, and certain determinations made at the partner
level. Accordingly, partnerships holding our common stock and the partners in such partnerships should consult their tax advisors regarding
the U.S. federal income tax consequences to them.
THIS
DISCUSSION IS FOR INFORMATIONAL PURPOSES ONLY AND IS NOT TAX ADVICE. INVESTORS SHOULD CONSULT THEIR TAX ADVISORS WITH RESPECT TO THE
APPLICATION OF THE U.S. FEDERAL INCOME TAX LAWS TO THEIR PARTICULAR SITUATIONS AS WELL AS ANY TAX CONSEQUENCES OF THE PURCHASE, OWNERSHIP,
AND DISPOSITION OF OUR COMMON STOCK ARISING UNDER THE U.S. FEDERAL ESTATE OR GIFT TAX LAWS OR UNDER THE LAWS OF ANY STATE, LOCAL, OR
NON-U.S. TAXING JURISDICTION OR UNDER ANY APPLICABLE INCOME TAX TREATY.
Definition
of a Non-U.S. Holder
For
purposes of this discussion, a “Non-U.S. Holder” is any beneficial owner of our common stock that is neither a “U.S.
person” nor an entity treated as a partnership for U.S. federal income tax purposes. A U.S. person is any person that, for U.S.
federal income tax purposes, is or is treated as any of the following:
| ● | an
individual who is a citizen or resident of the United States; |
| ● | a
corporation created or organized under the laws of the United States, any state thereof,
or the District of Columbia; |
| ● | an
estate, the income of which is subject to U.S. federal income tax regardless of its source;
or |
| ● | a
trust that (1) is subject to the primary supervision of a U.S. court and the control of one
or more “United States persons” (within the meaning of Section 7701(a)(30) of
the Code), or (2) has a valid election in effect to be treated as a United States person
for U.S. federal income tax purposes. |
Distributions
As
described in the section entitled “Dividend Policy,” we do not anticipate declaring or paying dividends to holders of our
common stock in the foreseeable future. However, if we do make distributions of cash or property on our common stock, such distributions
will constitute dividends for U.S. federal income tax purposes to the extent paid from our current or accumulated earnings and profits,
as determined under U.S. federal income tax principles. Amounts not treated as dividends for U.S. federal income tax purposes will constitute
a return of capital and first be applied against and reduce a Non-U.S. Holder’s adjusted tax basis in its common stock, but not
below zero. Any excess will be treated as capital gain and will be treated as described below under “—Sale or Other Taxable
Disposition.”
Subject
to the discussion below on effectively connected income, dividends paid to a Non-U.S. Holder will be subject to U.S. federal withholding
tax at a rate of 30% of the gross amount of the dividends (or such lower rate specified by an applicable income tax treaty, provided
the Non-U.S. Holder furnishes a valid IRS Form W-8BEN or W-8BEN-E (or other applicable documentation) certifying qualification for the
lower treaty rate). A Non-U.S. Holder that does not timely furnish the required documentation, but that qualifies for a reduced treaty
rate, may obtain a refund of any excess amounts withheld by timely filing an appropriate claim for refund with the IRS. Non-U.S. Holders
should consult their tax advisors regarding their entitlement to benefits under any applicable income tax treaty.
If
dividends paid to a Non-U.S. Holder are effectively connected with the Non-U.S. Holder’s conduct of a trade or business within
the United States (and, if required by an applicable income tax treaty, the Non-U.S. Holder maintains a permanent establishment in the
United States to which such dividends are attributable), the Non-U.S. Holder will be exempt from the U.S. federal withholding tax described
above. To claim the exemption, the Non-U.S. Holder must furnish to the applicable withholding agent a valid IRS Form W-8ECI, certifying
that the dividends are effectively connected with the Non-U.S. Holder’s conduct of a trade or business within the United States.
Any
such effectively connected dividends will be subject to U.S. federal income tax on a net income basis at the regular rates. A Non-U.S.
Holder that is a corporation also may be subject to a branch profits tax at a rate of 30% (or such lower rate specified by an applicable
income tax treaty) on such effectively connected dividends, as adjusted for certain items. Non-U.S. Holders should consult their tax
advisors regarding any applicable tax treaties that may provide for different rules.
Sale
or Other Taxable Disposition
A
Non-U.S. Holder will not be subject to U.S. federal income tax on any gain realized upon the sale or other taxable disposition of our
common stock unless:
| ● | the
gain is effectively connected with the Non-U.S. Holder’s conduct of a trade or business
within the United States (and, if required by an applicable income tax treaty, the Non-U.S.
Holder maintains a permanent establishment in the United States to which such gain is attributable); |
| ● | the
Non-U.S. Holder is a nonresident alien individual present in the United States for 183 days
or more during the taxable year of the disposition and certain other requirements are met;
or |
| ● | our
common stock constitutes a U.S. real property interest, or USRPI, by reason of our status
as a U.S. real property holding corporation, or USRPHC, for U.S. federal income tax purposes. |
Gain
described in the first bullet point above generally will be subject to U.S. federal income tax on a net income basis at the regular rates.
A Non-U.S. Holder that is a corporation also may be subject to a branch profits tax at a rate of 30% (or such lower rate specified by
an applicable income tax treaty) on such effectively connected gain, as adjusted for certain items.
A
Non-U.S. Holder described in the second bullet point above will be subject to U.S. federal income tax at a rate of 30% (or such lower
rate specified by an applicable income tax treaty) on gain realized upon the sale or other taxable disposition of our common stock, which
may be offset by U.S. source capital losses of the Non-U.S. Holder (even though the individual is not considered a resident of the United
States), provided the Non-U.S. Holder has timely filed U.S. federal income tax returns with respect to such losses.
With
respect to the third bullet point above, we believe we currently are not, and do not anticipate becoming, a USRPHC. Because the determination
of whether we are a USRPHC depends, however, on the fair market value of our USRPIs relative to the fair market value of our USRPIs and
our other business assets, there can be no assurance we currently are not a USRPHC or will not become one in the future. Even if we are
or were to become a USRPHC, gain arising from the sale or other taxable disposition of our common stock by a Non-U.S. Holder will not
be subject to U.S. federal income tax if our common stock is “regularly traded,” as defined by applicable Treasury Regulations,
on an established securities market and such Non-U.S. Holder owned, actually and constructively, 5% or less of our common stock throughout
the shorter of the five-year period ending on the date of the sale or other taxable disposition or the Non-U.S. Holder’s holding
period.
Non-U.S.
Holders should consult their tax advisors regarding potentially applicable income tax treaties that may provide for different rules.
Information
Reporting and Backup Withholding
Payments
of dividends on our common stock will not be subject to backup withholding, provided the applicable withholding agent does not have actual
knowledge or reason to know the holder is a United States person and the holder either certifies its non-U.S. status, such as by furnishing
a valid IRS Form W-8BEN, W-8BEN-E, or W-8ECI, or otherwise establishes an exemption. However, information returns are required to be
filed with the IRS in connection with any distributions on our common stock paid to the Non-U.S. Holder, regardless of whether such distributions
constitute dividends or whether any tax was actually withheld. In addition, proceeds of the sale or other taxable disposition of our
common stock within the United States or conducted through certain U.S.-related brokers generally will not be subject to backup withholding
or information reporting if the applicable withholding agent receives the certification described above and does not have actual knowledge
or reason to know that such holder is a United States person or the holder otherwise establishes an exemption. Proceeds of a disposition
of our common stock conducted through a non-U.S. office of a non-U.S. broker generally will not be subject to backup withholding or information
reporting.
Copies
of information returns that are filed with the IRS may also be made available under the provisions of an applicable treaty or agreement
to the tax authorities of the country in which the Non-U.S. Holder resides or is established.
Backup
withholding is not an additional tax. Any amounts withheld under the backup withholding rules may be allowed as a refund or a credit
against a Non-U.S. Holder’s U.S. federal income tax liability, provided the required information is timely furnished to the IRS.
Additional
Withholding Tax on Payments Made to Foreign Accounts
Withholding
taxes may be imposed under Sections 1471 to 1474 of the Code (such Sections commonly referred to as the Foreign Account Tax Compliance
Act, or FATCA) on certain types of payments made to non-U.S. financial institutions and certain other non-U.S. entities. Specifically,
a 30% withholding tax may be imposed on dividends on, or (subject to the proposed Treasury Regulations discussed below) gross proceeds
from the sale or other disposition of, our common stock paid to a “foreign financial institution” or a “non-financial
foreign entity” (each as defined in the Code), unless (1) the foreign financial institution undertakes certain diligence and reporting
obligations, (2) the non-financial foreign entity either certifies it does not have any “substantial United States owners”
(as defined in the Code) or furnishes identifying information regarding each substantial United States owner, or (3) the foreign financial
institution or non-financial foreign entity otherwise qualifies for an exemption from these rules. If the payee is a foreign financial
institution and is subject to the diligence and reporting requirements in (1) above, it must enter into an agreement with the U.S. Department
of the Treasury requiring, among other things, that it undertake to identify accounts held by certain “specified United States
persons” or “United States owned foreign entities” (each as defined in the Code), annually report certain information
about such accounts, and withhold 30% on certain payments to non-compliant foreign financial institutions and certain other account holders.
Foreign financial institutions located in jurisdictions that have an intergovernmental agreement with the United States governing FATCA
may be subject to different rules.
Under
the applicable Treasury Regulations and administrative guidance, withholding under FATCA generally applies to payments of dividends on
our common stock. While withholding under FATCA would have applied also to payments of gross proceeds from the sale or other disposition
of stock on or after January 1, 2019, proposed Treasury Regulations eliminate FATCA withholding on payments of gross proceeds entirely.
Taxpayers generally may rely on these proposed Treasury Regulations until final Treasury Regulations are issued.
Prospective
investors should consult their tax advisors regarding the potential application of withholding under FATCA to their investment in our
common stock.
UNDERWRITING
ThinkEquity
LLC is acting as representative of the underwriters of this offering. We have entered into an underwriting agreement dated with the
representative. Subject to the terms and conditions of the underwriting agreement, we have agreed to sell to each underwriter named below,
and each underwriter named below has severally agreed to purchase, at the public offering price less the underwriting discounts set forth
on the cover page of this prospectus, the number of shares of common stock and Pre-Funded Warrants listed next to its name in
the following table:
| Underwriter | |
Number
of Shares | |
|
Number of Pre-Funded Warrants |
|
| ThinkEquity
LLC | |
| | |
|
|
|
|
| Total | |
| | |
|
|
|
|
The
underwriters are committed to purchase all shares of common stock and Pre-Funded Warrants offered by us other than those covered
by the over-allotment option described below, if any are purchased. The obligations of the underwriters may be terminated upon the occurrence
of certain events specified in the underwriting agreement. Furthermore, pursuant to the underwriting agreement, the underwriters’
obligations are subject to customary conditions, representations and warranties contained in the underwriting agreement, such as receipt
by the underwriters of officers’ certificates and legal opinions.
The
underwriters are offering the shares of common stock and Pre-Funded Warrants subject to prior sale, when, as and if issued to
and accepted by them, subject to approval of legal matters by their counsel, and other conditions. The underwriters reserve the right
to withdraw, cancel or modify offers to the public and to reject orders in whole or in part.
The
underwriters propose to offer the shares of common stock and Pre-Funded Warrants offered by us to the public at the public offering
price set forth on the cover of the prospectus. After the shares of common stock and Pre-Funded Warrants are released for sale
to the public, the underwriters may change the offering price and other selling terms at various times.
Over-Allotment
Option
We
have granted the underwriters an over-allotment option. This option, which is exercisable for up to 45 days after the date of this prospectus,
permits the representative to purchase a maximum of 450,000 additional shares of common stock and/or Pre-Funded Warrants (15%
of the shares and/or Pre-Funded Warrants sold in this offering) from us to cover over-allotments, if any. If the representative
exercises all or part of this option, it will purchase shares and/or Pre-Funded Warrants covered by the option at the public offering
price per share that appears on the cover page of this prospectus, less the underwriting discount. If this option is exercised in full,
the total offering price to the public will be $7.1 million and the total net proceeds, before expenses, to us will be $6.0
million.
Discount
The
underwriters propose initially to offer the shares of common stock and/or Pre-Funded Warrants to the public at the public
offering price set forth on the cover page of this prospectus and to dealers at that price less a concession not in excess of $ per share of common stock. If all of the shares of common stock and/or Pre-Funded Warrants offered by
us are not sold at the public offering price, the underwriters may change the offering price and other selling terms by means of a
supplement to this prospectus.
The
following table shows the public offering price, underwriting discounts and proceeds, before expenses, to us. The information assumes
either no exercise or full exercise by the underwriters of their over-allotment option.
| | |
Per Share | | |
Per Pre-Funded Warrant | | |
Total Without Over-Allotment Option | | |
Total With Over-Allotment Option | |
| Public offering price | |
$ | | | |
$ | | | |
| | | |
| | |
| Underwriting discount (7.5%) | |
$ | | | |
$ | | | |
| | | |
| | |
| Proceeds, before expense, to us | |
$ | | | |
$ | | | |
| | | |
| | |
We
have agreed to pay a non-accountable expense allowance to the underwriters equal to 1% of the gross proceeds received in this offering
(excluding proceeds received from exercise of the underwriters’ over-allotment option).
We
will pay an expense deposit of $35,000 to the representative
for out-of-pocket-accountable expenses, which will be returned to us to the extent such out-of-pocket accountable expenses are not actually
incurred in accordance with FINRA Rule 5110(g)(4)(A).
In
addition, we have agreed to reimburse the representative for (i) fees and expenses of legal counsel to the underwriters in an amount
not to exceed $125,000; (ii) fees and expenses related to the use of Ipreo’s book building, prospectus tracking and compliance
software for the offering in the amount of $21,000; (iii) all fees, expenses and disbursements relating to the registration, qualification
or exemption of such shares under the securities laws of such foreign jurisdictions as the representative may reasonably designate; (iv)
all fees, expenses and disbursements relating to the registration, qualification or exemption of such shares under the “blue sky”
securities laws of such states, if applicable, and other jurisdictions as the representative may reasonably designate; and (v)
up to $10,000 for actual accountable “road show” expenses.
We
estimate that the total expenses of the offering payable by us, excluding the total underwriting discount and non-accountable expense
allowance, will be approximately $500,000.
Representative’s
Warrants
We
have agreed to issue to the representative or its designees warrants to purchase up to a total of 120,000 (or 138,000 if
the over-allotment option is exercised in full) shares of our common stock (4% of the aggregate number of shares of common stock and/or
Pre-Funded Warrants sold in this offering), or the Representative’s Warrants. The Representative’s Warrants will be exercisable
at a per share exercise price equal to 125% of the public offering price per share of the shares of common stock sold in this offering.
The Representative’s Warrants are exercisable at any time, from time to time, in whole or in part, during the four and one half
year period commencing 180 days from the commencement of sales of the securities in this offering.
The
Representative’s Warrants and the shares of common stock underlying the Representative’s Warrants have been deemed compensation
by FINRA and are, therefore, subject to a 180-day lock-up pursuant to FINRA Rule 5110(e)(1). The representative or (permitted assignees
under Rule 5110(e)(2)) will not sell, transfer, assign, pledge, or hypothecate the Representative’s Warrants or the securities
underlying the Representative’s Warrants, nor will the representative engage in any hedging, short sale, derivative, put, or call
transaction that would result in the effective economic disposition of the Representative’s Warrants or the underlying shares of
common stock for a period of 180 days from the effective date of the registration statement. Additionally, the Representative’s
Warrants may not be sold, transferred, assigned, pledged, or hypothecated for a 180-day period following the effective date of the registration
statement, except to any underwriter and selected dealer participating in the offering and their bona fide officers or partners. The
Representative’s Warrants will provide for adjustment in the number and price of the Representative’s Warrants and the shares
of common stock underlying the Representative’s Warrants in the event of recapitalization, merger, stock split, or other structural
transaction, or a future financing undertaken by us. The Representative’s Warrants will provide for registration rights (including
a one-time demand registration right and unlimited piggyback rights) consistent with FINRA Rule 5110.05. The demand for registration
may be made at any time beginning on the initial exercise date of the Representative’s Warrants and expiring on the fifth anniversary
of the effective date of this registration statement in accordance with FINRA Rule 5110(g)(8)(C). In addition to the one-time demand
registration right, the Representative’s Warrants shall have unlimited piggyback rights, for a period of no more than two years
from the initial exercise date of the Representative’s Warrants in accordance with FINRA Rule 5110(g)(8)(D). The Representative’s
Warrants will also provide for customary anti-dilution provisions (for stock dividends and splits and recapitalizations) consistent with
FINRA Rule 5110, and further, the number of shares underlying the Representative’s Warrants shall be reduced if necessary to comply
with FINRA rules and regulations.
Discretionary
Accounts
The
underwriters do not intend to confirm sales of the securities offered hereby to any accounts over which they have discretionary authority.
Lock-Up
Agreements
Each
of our directors and officers have agreed, subject to certain exceptions, not to offer, pledge, sell, contract to sell, grant, lend,
or otherwise transfer or dispose of, directly or indirectly, or enter into any swap or other arrangement that transfers to another, in
whole or in part, any of the economic consequences of ownership of any shares of our capital stock or any securities convertible into
or exercisable or exchangeable for shares of our common stock, for a period of six (6) months after the date of this prospectus, without
the prior written consent of the representative. We, subject to certain exceptions, have agreed, subject to certain exceptions, not offer,
pledge, sell, contract to sell, grant, lend, or otherwise transfer or dispose of, directly or indirectly, or enter into any swap or other
arrangement that transfers to another, in whole or in part, any of the economic consequences of ownership of any shares of our capital
stock or any securities convertible into or exercisable or exchangeable for shares of our common stock, for a period of three (3) months
after the date of this prospectus, without the prior written consent of the representative.
Additionally,
we agreed that for a period of six (6) months after this offering we will not directly or indirectly in any “at-the-market,”
continuous equity, equity lines, or variable rate transaction, offer to sell, sell, contract to sell, grant any option to sell or otherwise
dispose of shares of our capital stock or any securities convertible into or exercisable or exchangeable for our shares of capital stock,
without the prior written consent of ThinkEquity.
Following
the expiration of the applicable lock-up period, all of the issued and outstanding shares of our common stock will be eligible for future
sale, subject to the applicable volume, manner of sale, holding period, and other limitations of Rule 144.
Right
of First Refusal
In
connection with our IPO, we agreed that for a period of fifteen (15) months from the closing of the offering, we will grant the representative
an irrevocable right of first refusal to act as sole investment banker, sole book-runner, sole financial advisor, sole underwriter and/or
sole placement agent, at the representative’s sole discretion, for each and every future public and private equity and debt offering,
including all equity linked financings, during such fifteen (15) month period for us, or any successor to or any subsidiary of us, on
terms customary to the representative. We also agreed that the representative has the sole right to determine whether or not any other
broker dealer shall have the right to participate in any such offering and the economic terms of any such participation.
In
connection with this Offering, contingent upon securing a minimum of $15 million in offering proceeds, the Underwriting Agreement will
provide for a six (6) month extension of the right of first refusal initially granted to the representative during our IPO.
Indemnification
To
the extent permitted by law, we have agreed to indemnify the underwriters and its affiliates, stockholders, directors, officers, employees,
members and controlling persons against certain liabilities, including liabilities under the Securities Act, and to contribute to payments
that the underwriters may be required to make for these liabilities.
Electronic
Offer, Sale and Distribution of Shares
A
prospectus in electronic format may be made available on the websites maintained by one or more underwriters or selling group members,
if any, participating in this offering and one or more of the underwriters participating in this offering may distribute prospectuses
electronically. The representative may agree to allocate a number of shares to underwriters and selling group members for sale to their
online brokerage account holders. Internet distributions will be allocated by the underwriters and selling group members that will make
internet distributions on the same basis as other allocations. Other than the prospectus in electronic format, the information on the
underwriters’ websites is not part of, nor incorporated by reference into, this prospectus or the registration statement of which
this prospectus forms a part, has not been approved or endorsed by us or any underwriter in its capacity as underwriter, and should not
be relied upon by investors.
Stabilization
In
connection with this offering, the underwriters may engage in stabilizing transactions, over-allotment transactions, syndicate-covering
transactions, penalty bids and purchases to cover positions created by short sales.
Stabilizing
transactions permit bids to purchase securities so long as the stabilizing bids do not exceed a specified maximum and are engaged in
for the purpose of preventing or retarding a decline in the market price of the securities while the offering is in progress.
Over-allotment
transactions involve sales by the underwriters of securities in excess of the number of securities that underwriters are obligated to
purchase. This creates a syndicate short position which may be either a covered short position or a naked short position. In a covered
short position, the number of securities over-allotted by the underwriters is not greater than the number of securities that they may
purchase in the over-allotment option. In a naked short position, the number of securities involved is greater than the number of securities
in the over-allotment option. The underwriters may close out any short position by exercising their over-allotment option and/or purchasing
securities in the open market.
Syndicate
covering transactions involve purchases of securities in the open market after the distribution has been completed in order to cover
syndicate short positions. In determining the source of securities to close out the short position, the underwriters will consider, among
other things, the price of securities available for purchase in the open market as compared with the price at which they may purchase
securities through exercise of the over-allotment option. If the underwriters sell more securities than could be covered by exercise
of the over-allotment option and, therefore, have a naked short position, the position can be closed out only by buying securities in
the open market. A naked short position is more likely to be created if the underwriters are concerned that after pricing there could
be downward pressure on the price of the securities in the open market that could adversely affect investors who purchase in the offering.
Penalty
bids permit the representative to reclaim a selling concession from a syndicate member when the securities originally sold by that syndicate
member are purchased in stabilizing or syndicate covering transactions to cover syndicate short positions.
These
stabilizing transactions, syndicate covering transactions and penalty bids may have the effect of raising or maintaining the market price
of our securities or preventing or retarding a decline in the market price of our securities. As a result, the price of our securities
in the open market may be higher than it would otherwise be in the absence of these transactions. Neither we nor the underwriters make
any representation or prediction as to the effect that the transactions described above may have on the price of our securities. These
transactions may be effected on the NYSE American, in the over-the-counter market or otherwise and, if commenced, may be discontinued
at any time.
Passive
Market Making
In
connection with this offering, underwriters and selling group members may engage in passive market making transactions in our common
stock on the NYSE American or on the OTCQB in accordance with Rule 103 of Regulation M under the Exchange Act, during a period before
the commencement of offers or sales of the securities and extending through the completion of the distribution. A passive market maker
must display its bid at a price not in excess of the highest independent bid of that security. However, if all independent bids are lowered
below the passive market maker’s bid, then that bid must then be lowered when specified purchase limits are exceeded.
Other
Relationships
Certain
of the underwriters and their affiliates may provide in the future, various advisory, investment and commercial banking and other services
to us in the ordinary course of business, for which they may receive customary fees and commissions. However, we have not yet had, and
have no present arrangements with any of the underwriters for any further services.
Offer
Restrictions Outside the United States
Other
than in the United States, no action has been taken by us or the underwriters that would permit a public offering of the securities offered
by this prospectus in any jurisdiction where action for that purpose is required. The securities offered by this prospectus may not be
offered or sold, directly or indirectly, nor may this prospectus or any other offering material or advertisements in connection with
the offer and sale of any such securities be distributed or published in any jurisdiction, except under circumstances that will result
in compliance with the applicable rules and regulations of that jurisdiction. Persons into whose possession this prospectus comes are
advised to inform themselves about and to observe any restrictions relating to this offering and the distribution of this prospectus.
This prospectus does not constitute an offer to sell or a solicitation of an offer to buy any securities offered by this prospectus in
any jurisdiction in which such an offer or a solicitation is unlawful.
Australia
This
prospectus is not a disclosure document under Chapter 6D of the Australian Corporations Act, has not been lodged with the Australian
Securities and Investments Commission and does not purport to include the information required of a disclosure document under Chapter
6D of the Australian Corporations Act. Accordingly, (i) the offer of the securities under this prospectus is only made to persons to
whom it is lawful to offer the securities without disclosure under Chapter 6D of the Australian Corporations Act under one or more exemptions
set out in section 708 of the Australian Corporations Act, (ii) this prospectus is made available in Australia only to those persons
as set forth in clause (i) above, and (iii) the offeree must be sent a notice stating in substance that by accepting this offer, the
offeree represents that the offeree is such a person as set forth in clause (i) above, and, unless permitted under the Australian Corporations
Act, agrees not to sell or offer for sale within Australia any of the securities sold to the offeree within 12 months after its transfer
to the offeree under this prospectus.
China
The
information in this document does not constitute a public offer of the securities, whether by way of sale or subscription, in the People’s
Republic of China (excluding, for purposes of this paragraph, Hong Kong Special Administrative Region, Macau Special Administrative Region
and Taiwan). The securities may not be offered or sold directly or indirectly in the PRC to legal or natural persons other than directly
to “qualified domestic institutional investors.”
European
Economic Area—Belgium, Germany, Luxembourg and Netherlands
The
information in this document has been prepared on the basis that all offers of securities will be made pursuant to an exemption under
the Directive 2003/71/EC (“Prospectus Directive”), as implemented in Member States of the European Economic Area (each, a
“Relevant Member State”), from the requirement to produce a prospectus for offers of securities.
An
offer to the public of securities has not been made, and may not be made, in a Relevant Member State except pursuant to one of the following
exemptions under the Prospectus Directive as implemented in that Relevant Member State:
| |
● |
to
legal entities that are authorized or regulated to operate in the financial markets or, if not so authorized or regulated, whose
corporate purpose is solely to invest in securities; |
| |
|
|
| |
● |
to
any legal entity that has two or more of (i) an average of at least 250 employees during its last fiscal year; (ii) a total balance
sheet of more than €43,000,000 (as shown on its last annual unconsolidated or consolidated financial statements) and (iii) an
annual net turnover of more than €50,000,000 (as shown on its last annual unconsolidated or consolidated financial statements); |
| |
● |
to
fewer than 100 natural or legal persons (other than qualified investors within the meaning of Article 2(1)(e) of the Prospectus Directive)
subject to obtaining the prior consent of the Company or any underwriter for any such offer; or |
| |
|
|
| |
● |
in
any other circumstances falling within Article 3(2) of the Prospectus Directive, provided that no such offer of securities shall
result in a requirement for the publication by the Company of a prospectus pursuant to Article 3 of the Prospectus Directive. |
France
This
document is not being distributed in the context of a public offering of financial securities (offre au public de titres financiers)
in France within the meaning of Article L.411-1 of the French Monetary and Financial Code (Code monétaire et financier) and Articles
211-1 et seq. of the General Regulation of the French Autorité des marchés financiers (“AMF”). The securities
have not been offered or sold and will not be offered or sold, directly or indirectly, to the public in France.
This
document and any other offering material relating to the securities have not been, and will not be, submitted to the AMF for approval
in France and, accordingly, may not be distributed or caused to distributed, directly or indirectly, to the public in France.
Such
offers, sales and distributions have been and shall only be made in France to (i) qualified investors (investisseurs qualifiés)
acting for their own account, as defined in and in accordance with Articles L.411-2-II-2 and D.411-1 to D.411-3, D. 744-1, D.754-1 and
D.764-1 of the French Monetary and Financial Code and any implementing regulation and/or (ii) a restricted number of non-qualified investors
(cercle restreint d’investisseurs) acting for their own account, as defined in and in accordance with Articles L.411-2-II-2 and
D.411-4, D.744-1, D.754-1 and D.764-1 of the French Monetary and Financial Code and any implementing regulation.
Pursuant
to Article 211-3 of the General Regulation of the AMF, investors in France are informed that the securities cannot be distributed (directly
or indirectly) to the public by the investors otherwise than in accordance with Articles L.411-1, L.411-2, L.412-1 and L.621-8 to L.621-8-3
of the French Monetary and Financial Code.
Ireland
The
information in this document does not constitute a prospectus under any Irish laws or regulations and this document has not been filed
with or approved by any Irish regulatory authority as the information has not been prepared in the context of a public offering of securities
in Ireland within the meaning of the Irish Prospectus (Directive 2003/71/EC) Regulations 2005 (the “Prospectus Regulations”).
The securities have not been offered or sold, and will not be offered, sold or delivered directly or indirectly in Ireland by way of
a public offering, except to (i) qualified investors as defined in Regulation 2(l) of the Prospectus Regulations and (ii) fewer than
100 natural or legal persons who are not qualified investors.
Israel
The
securities offered by this prospectus have not been approved or disapproved by the Israeli Securities Authority (the ISA), or ISA, nor
have such securities been registered for sale in Israel. The shares may not be offered or sold, directly or indirectly, to the public
in Israel, absent the publication of a prospectus. The ISA has not issued permits, approvals or licenses in connection with this offering
or publishing the prospectus; nor has it authenticated the details included herein, confirmed their reliability or completeness, or rendered
an opinion as to the quality of the securities being offered. Any resale in Israel, directly or indirectly, to the public of the securities
offered by this prospectus is subject to restrictions on transferability and must be effected only in compliance with the Israeli securities
laws and regulations.
Italy
The
offering of the securities in the Republic of Italy has not been authorized by the Italian Securities and Exchange Commission (Commissione
Nazionale per le Societa e la Borsa, or CONSOB), pursuant to the Italian securities legislation and, accordingly, no offering material
relating to the securities may be distributed in Italy and such securities may not be offered or sold in Italy in a public offer within
the meaning of Article 1.1(t) of Legislative Decree No. 58 of 24 February 1998 (“Decree No. 58”), other than:
| |
● |
to
Italian qualified investors (“Qualified Investors”), as defined in Article 100 of Decree no. 58 by reference to Article
34-ter of CONSOB Regulation no. 11971 of 14 May 1999, as amended (“Regulation no. 1197l”); and |
| |
|
|
| |
● |
in
other circumstances that are exempt from the rules on public offer pursuant to Article 100 of Decree No. 58 and Regulation no. 1197l. |
Any
offer, sale or delivery of the securities or distribution of any offer document relating to the securities in Italy (excluding placements
where a Qualified Investor solicits an offer from the issuer) under the paragraphs above must be:
| |
● |
made
by investment firms, banks or financial intermediaries permitted to conduct such activities in Italy in accordance with Legislative
Decree No. 385 of 1 September 1993, as amended, Decree No. 58, CONSOB Regulation No. 16190 of 29 October 2007, and any other applicable
laws; and |
| |
|
|
| |
● |
in
compliance with all relevant Italian securities, tax and exchange controls and any other applicable laws. |
Any
subsequent distribution of the securities in Italy must be made in compliance with the public offer and prospectus requirement rules
provided under Decree No. 58 and the Regulation No. 11971 as amended, unless an exception from those rules applies. Failure to comply
with such rules may result in the sale of such securities being declared null and void and in the liability of the entity transferring
the securities for any damages suffered by the investors.
Japan
The
securities have not been and will not be registered under Article 4, paragraph 1 of the Financial Instruments and Exchange Law of Japan
(Law No. 25 of 1948), as amended (the “FIEL”) pursuant to an exemption from the registration requirements applicable to a
private placement of securities to Qualified Institutional Investors (as defined in and in accordance with Article 2, paragraph 3 of
the FIEL and the regulations promulgated thereunder). Accordingly, the securities may not be offered or sold, directly or indirectly,
in Japan or to, or for the benefit of, any resident of Japan other than Qualified Institutional Investors. Any Qualified Institutional
Investor who acquires securities may not resell them to any person in Japan that is not a Qualified Institutional Investor, and acquisition
by any such person of securities is conditional upon the execution of an agreement to that effect.
Portugal
This
document is not being distributed in the context of a public offer of financial securities (oferta pública de valores mobiliários)
in Portugal, within the meaning of Article 109 of the Portuguese Securities Code (Código dos Valores Mobiliários). The
securities have not been offered or sold and will not be offered or sold, directly or indirectly, to the public in Portugal. This document
and any other offering material relating to the securities have not been, and will not be, submitted to the Portuguese Securities Market
Commission (Comissăo do Mercado de Valores Mobiliários) for approval in Portugal and, accordingly, may not be distributed
or caused to distributed, directly or indirectly, to the public in Portugal, other than under circumstances that are deemed not to qualify
as a public offer under the Portuguese Securities Code. Such offers, sales and distributions of securities in Portugal are limited to
persons who are “qualified investors” (as defined in the Portuguese Securities Code). Only such investors may receive this
document and they may not distribute it or the information contained in it to any other person.
Sweden
This
document has not been, and will not be, registered with or approved by Finansinspektionen (the Swedish Financial Supervisory Authority).
Accordingly, this document may not be made available, nor may the securities be offered for sale in Sweden, other than under circumstances
that are deemed not to require a prospectus under the Swedish Financial Instruments Trading Act (1991:980) (Sw. lag (1991:980) om handel
med finansiella instrument). Any offering of securities in Sweden is limited to persons who are “qualified investors” (as
defined in the Financial Instruments Trading Act). Only such investors may receive this document and they may not distribute it or the
information contained in it to any other person.
Switzerland
The
securities may not be publicly offered in Switzerland and will not be listed on the SIX Swiss Exchange (“SIX”) or on any
other stock exchange or regulated trading facility in Switzerland. This document has been prepared without regard to the disclosure standards
for issuance prospectuses under art. 652a or art. 1156 of the Swiss Code of Obligations or the disclosure standards for listing prospectuses
under art. 27 ff. of the SIX Listing Rules or the listing rules of any other stock exchange or regulated trading facility in Switzerland.
Neither this document nor any other offering material relating to the securities may be publicly distributed or otherwise made publicly
available in Switzerland.
Neither
this document nor any other offering material relating to the securities have been or will be filed with or approved by any Swiss regulatory
authority. In particular, this document will not be filed with, and the offer of securities will not be supervised by, the Swiss Financial
Market Supervisory Authority (FINMA).
This
document is personal to the recipient only and not for general circulation in Switzerland.
United
Arab Emirates
Neither
this document nor the securities have been approved, disapproved or passed on in any way by the Central Bank of the United Arab Emirates
or any other governmental authority in the United Arab Emirates, nor has the Company received authorization or licensing from the Central
Bank of the United Arab Emirates or any other governmental authority in the United Arab Emirates to market or sell the securities within
the United Arab Emirates. This document does not constitute and may not be used for the purpose of an offer or invitation. No services
relating to the securities, including the receipt of applications and/or the allotment or redemption of such shares, may be rendered
within the United Arab Emirates by the Company.
No
offer or invitation to subscribe for securities is valid or permitted in the Dubai International Financial Centre.
United
Kingdom
Neither
the information in this document nor any other document relating to the offer has been delivered for approval to the Financial Services
Authority in the United Kingdom and no prospectus (within the meaning of section 85 of the Financial Services and Markets Act 2000, as
amended (“FSMA”)) has been published or is intended to be published in respect of the securities. This document is issued
on a confidential basis to “qualified investors” (within the meaning of section 86(7) of FSMA) in the United Kingdom, and
the securities may not be offered or sold in the United Kingdom by means of this document, any accompanying letter or any other document,
except in circumstances which do not require the publication of a prospectus pursuant to section 86(1) FSMA. This document should not
be distributed, published or reproduced, in whole or in part, nor may its contents be disclosed by recipients to any other person in
the United Kingdom.
Any
invitation or inducement to engage in investment activity (within the meaning of section 21 of FSMA) received in connection with the
issue or sale of the securities has only been communicated or caused to be communicated and will only be communicated or caused to be
communicated in the United Kingdom in circumstances in which section 21(1) of FSMA does not apply to the Company. In the United Kingdom,
this document is being distributed only to, and is directed at, persons (i) who have professional experience in matters relating to investments
falling within Article 19(5) (investment professionals) of the Financial Services and Markets Act 2000 (Financial Promotions) Order 2005
(“FPO”), (ii) who fall within the categories of persons referred to in Article 49(2)(a) to (d) (high net worth companies,
unincorporated associations, etc.) of the FPO or (iii) to whom it may otherwise be lawfully communicated (together “relevant persons”).
The investments to which this document relates are available only to, and any invitation, offer or agreement to purchase will be engaged
in only with, relevant persons. Any person who is not a relevant person should not act or rely on this document or any of its contents.
Canada
The
securities may be sold in Canada only to purchasers purchasing, or deemed to be purchasing, as principal that are accredited investors,
as defined in National Instrument 45-106 Prospectus Exemptions or subsection 73.3(1) of the Securities Act (Ontario), and are permitted
clients, as defined in National Instrument 31-103 Registration Requirements, Exemptions and Ongoing Registrant Obligations. Any resale
of the securities must be made in accordance with an exemption from, or in a transaction not subject to, the prospectus requirements
of applicable securities laws. Securities legislation in certain provinces or territories of Canada may provide a purchaser with remedies
for rescission or damages if this prospectus (including any amendment thereto) contains a misrepresentation, provided that the remedies
for rescission or damages are exercised by the purchaser within the time limit prescribed by the securities legislation of the purchaser’s
province or territory. The purchaser should refer to any applicable provisions of the securities legislation of the purchaser’s
province or territory for particulars of these rights or consult with a legal advisor. Pursuant to section 3A.3 of National Instrument
33-105 Underwriting Conflicts (NI 33-105), the underwriters are not required to comply with the disclosure requirements of NI33-105 regarding
underwriter conflicts of interest in connection with this offering.
LEGAL
MATTERS
Greenberg
Traurig, LLP, Irvine, California, will pass upon the validity of the shares of common stock and Pre-Funded Warrants offered hereby.
Venable LLP, New York, New York, has acted as counsel for the underwriters in connection with certain legal matters related to this offering.
EXPERTS
The
financial statements as of and for the fiscal years ended December 31, 2022 and 2021 included in this prospectus have been so included
in reliance on the report of Grassi & Co., CPAs, P.C., an independent registered public accounting firm, given on the authority of
said firm as experts in auditing and accounting.
WHERE
YOU CAN FIND MORE INFORMATION
We
have filed with the SEC a registration statement on Form S-1 under the Securities Act with respect to the shares of common stock offered
by this prospectus. This prospectus, which constitutes a part of the registration statement, does not contain all the information set
forth in the registration statement, some of which is contained in exhibits to the registration statement as permitted by the rules and
regulations of the SEC. For further information with respect to us and our shares of common stock, we refer you to the registration statement,
including the exhibits filed as a part of the registration statement. Statements contained in this prospectus concerning the contents
of any contract or any other document is not necessarily complete. If a contract or document has been filed as an exhibit to the registration
statement, please see the copy of the contract or document that has been filed. Each statement is this prospectus relating to a contract
or document filed as an exhibit is qualified in all respects by the filed exhibit. The SEC maintains an Internet website that contains
reports, proxy statements and other information about issuers, like us, that file electronically with the SEC. The address of that website
is www.sec.gov.
In
addition, we file annual, quarterly and current reports and proxy statements and other information with the SEC. Our SEC filings are
available on the SEC’s website at www.sec.gov. These filings are also available free of charge to the public on, or accessible
through, our corporate website at www.azitrainc.com. Our website and the information contained on, or that can be accessed through, our
website is not deemed to be incorporated by reference in, and is not considered part of, this prospectus. You should not rely on any
such information in making your decision whether to purchase our common stock.
We
have not authorized anyone to give you any information or to make any representations about us or the transactions we discuss in this
prospectus other than those contained in this prospectus. If you are given any information or representations about these matters that
is not discussed in this prospectus, you must not rely on that information. This prospectus is not an offer to sell or a solicitation
of an offer to buy securities anywhere or to anyone where or to whom we are not permitted to offer or sell securities under applicable
law.
AZITRA,
INC.
INDEX
TO FINANCIAL STATEMENTS
| Report
of Independent Registered Public Accounting Firm (PCAOB ID 606) |
F-1 |
| Balance
Sheets as of December 31, 2022 and December 31, 2021 |
F-2 |
| Statements
of Operations for the years ended December 31, 2022 and 2021 |
F-3 |
| Statements
of Changes in Stockholders’ Equity (Deficit) for the years ended December 31, 2022 and 2021 |
F-4 |
| Statements
of Cash Flows for the years ended December 31, 2022 and 2021 |
F-5 |
| Notes
to Financial Statements |
F-6 |
| |
|
| Balance
Sheets as of September 30, 2023 (Unaudited) and December 31, 2022 |
F-24 |
| Statements
of Operations for the nine months ended September 30, 2023 and 2022 |
F-25 |
| Statements
of Stockholders’ Equity for the nine months ended September 30, 2023 and 2022 (Unaudited) |
F-26 |
| Statements
of Cash Flows for the nine months ended September 30, 2023 and 2022 |
F-27 |
| Notes
to Unaudited Financial Statements |
F-28 |
REPORT
OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To
the Board of Directors and Stockholders of
Azitra,
Inc.
Branford,
CT
Opinion
on the Financial Statements
We
have audited the accompanying balance sheets of Azitra, Inc. (the “Company”) as of December 31, 2022 and 2021, and the related
statements of operations, stockholders’ equity, and cash flows for the years then ended, and the related notes (collectively referred
to as the financial statements). In our opinion, the financial statements present fairly, in all material respects, the financial position
of the Company as of December 31, 2022 and 2021, and the results of its operations and its cash flows for the years then ended, in conformity
with accounting principles generally accepted in the United States of America.
Substantial
Doubt Regarding the Company’s Ability to Continue as a Going Concern
The
accompanying financial statements have been prepared assuming that Azitra, Inc., will continue as a going concern. As discussed in Note
1 to the financial statements, the Company’s significant operating losses raise substantial doubt about its ability to continue
as a going concern. Management’s evaluation of the events and conditions, and management’s plans regarding those matters,
are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis
for Opinion
These
financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s
financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board
(United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities
laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We
conducted our audits in accordance with the standards of the PCAOB and in accordance with auditing standards generally accepted in the
United States of America. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the
financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were
we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an
understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of
the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our
audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error
or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding
the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant
estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits
provide a reasonable basis for our opinion.
/s/
Grassi & CO., CPAs, P.C.
We
have served as the Company’s auditor since 2022.
Jericho,
New York
February
20, 2023, except for Notes 9, 10, 11, 14, and 19, as to which the date is June 13, 2023.
AZITRA,
INC.
Balance
Sheets
| | |
December
31, | |
| | |
2022 | | |
2021 | |
| Assets | |
| | | |
| | |
| Current
assets: | |
| | | |
| | |
| Cash
and cash equivalents | |
$ | 3,492,656 | | |
$ | 8,044,262 | |
| Accounts
receivable | |
| 182,820 | | |
| 160,867 | |
| Tax credits
receivable | |
| 69,666 | | |
| 173,311 | |
| Income
tax receivable | |
| 13,722 | | |
| - | |
| Deferred
offering costs | |
| 216,886 | | |
| - | |
| Prepaid
expenses | |
| 160,133 | | |
| 110,289 | |
| Total current assets | |
| 4,135,883 | | |
| 8,488,729 | |
| | |
| | | |
| | |
| Property and equipment, net | |
| 846,958 | | |
| 946,681 | |
| | |
| | | |
| | |
| Other assets | |
| | | |
| | |
| Other
assets | |
| 47,507 | | |
| 48,201 | |
| Operating
lease right-of-use asset | |
| 1,116,697 | | |
| - | |
| Intangible
assets, net | |
| 219,567 | | |
| 97,693 | |
| Deferred
patent costs | |
| 800,831 | | |
| 620,029 | |
| Total
other assets | |
| 2,184,602 | | |
| 765,923 | |
| | |
| | | |
| | |
| Total
assets | |
$ | 7,167,443 | | |
$ | 10,201,333 | |
| | |
| | | |
| | |
| Liabilities,
convertible preferred stock, and stockholders’ deficit | |
| | | |
| | |
| Current liabilities: | |
| | | |
| | |
| Accounts
payable | |
$ | 784,687 | | |
$ | 717,800 | |
| Current
operating lease liability | |
| 287,384 | | |
| - | |
| Income
tax payable | |
| - | | |
| 1,302 | |
| Accrued
expenses | |
| 993,961 | | |
| 475,485 | |
| Contract
liabilities | |
| 156,000 | | |
| 15,000 | |
| Total current liabilities | |
| 2,222,032 | | |
| 1,209,587 | |
| | |
| | | |
| | |
| Long-term
operating lease liability | |
| 840,896 | | |
| - | |
| Warrant
liability | |
| 70,283 | | |
| 71,104 | |
| Convertible
notes payable, net | |
| 6,600,000 | | |
| 992,019 | |
| Total liabilities | |
| 9,733,211 | | |
| 2,272,710 | |
| | |
| | | |
| | |
| Convertible preferred stock: | |
| | | |
| | |
| Series A convertible preferred
stock; $0.0001 par value; 205,385 shares authorized at December 31, 2022 and 2021; 205,385 shares issued and outstanding at December
31, 2022 and 2021; liquidation value of $3,337,506 as of December 31, 2022 and 2021 | |
| 3,272,944 | | |
| 3,272,944 | |
| Series A-1 convertible preferred
stock; $0.0001 par value; 380,657 shares authorized at December 31, 2022 and 2021; 380,657 shares issued and outstanding at December
31, 2022 and 2021; liquidation value of $14,274,638 as of December 31, 2022 and 2021 | |
| 14,100,533 | | |
| 14,100,533 | |
| Series
B convertible preferred stock; $0.0001 par value; 851,108 and 392,000 shares authorized at December 31, 2022 and 2021, respectively;
391,303 shares issued and outstanding at December 31, 2022 and 2021; liquidation value of $17,000,159 as of December 31, 2022 and
2021 | |
| 16,321,065 | | |
| 16,321,065 | |
| | |
| | | |
| | |
| Stockholders’ deficit: | |
| | | |
| | |
| Common
stock; $0.0001 par value; 100,000,000 authorized at December 31, 2022 and 2021, respectively; 1,043,991 and 1,043,103 shares issued
and outstanding at December 31, 2022 and 2021, respectively | |
| 104 | | |
| 104 | |
| Additional
paid-in capital | |
| 1,054,138 | | |
| 868,163 | |
| Accumulated
deficit | |
| (37,314,552 | ) | |
| (26,634,186 | ) |
| Total
stockholders’ deficit | |
| (36,260,310 | ) | |
| (25,765,919 | ) |
| | |
| | | |
| | |
| Total
liabilities, convertible preferred stock, and stockholders’ deficit | |
$ | 7,167,443 | | |
$ | 10,201,333 | |
See
accompanying notes
AZITRA,
INC.
Statements
of Operations
| | |
Years
ended December 31, | |
| | |
2022 | | |
2021 | |
| Service
revenue - related party | |
$ | 284,000 | | |
$ | 110,000 | |
| Total revenue | |
| 284,000 | | |
| 110,000 | |
| | |
| | | |
| | |
| Operating expenses: | |
| | | |
| | |
| General
and administrative | |
| 3,639,666 | | |
| 3,951,352 | |
| Research
and development | |
| 6,097,938 | | |
| 5,380,102 | |
| Total
operating expenses | |
| 9,737,604 | | |
| 9,331,454 | |
| | |
| | | |
| | |
| Loss from operations | |
| (9,453,604 | ) | |
| (9,221,454 | ) |
| | |
| | | |
| | |
| Other income (expense): | |
| | | |
| | |
| Interest
income | |
| 4,818 | | |
| 8,759 | |
| Interest
expense | |
| (251,891 | ) | |
| (66,968 | ) |
| Other
income | |
| 65,849 | | |
| 112,141 | |
| Employee
retention credit | |
| 229,813 | | |
| — | |
| Forgiveness
of Payroll Protection Program loan | |
| — | | |
| 232,506 | |
| Change
in fair value of convertible note | |
| (1,250,000 | ) | |
| — | |
| Other
expense | |
| (25,351 | ) | |
| (4,659 | ) |
| Total
other income (expense) | |
| (1,226,762 | ) | |
| 281,779 | |
| | |
| | | |
| | |
| Net loss before income taxes | |
| (10,680,366 | ) | |
| (8,939,675 | ) |
| | |
| | | |
| | |
| Income tax benefit (expense) | |
| | | |
| | |
| | |
| | | |
| | |
| Net loss | |
$ | (10,680,366 | ) | |
$ | (8,939,675 | ) |
| Dividends
on preferred stock | |
| (2,768,984 | ) | |
| (2,768,984 | ) |
| Net
loss attributable to common shareholders | |
$ | (13,449,350 | ) | |
$ | (11,708,659 | ) |
| | |
| | | |
| | |
| Net loss per share, basic
and diluted | |
$ | (12.74 | ) | |
$ | (11.20 | ) |
| Weighted average common stock
outstanding, basic and diluted | |
| 1,055,457 | | |
| 1,045,766 | |
See
accompanying notes
AZITRA,
INC.
Statements
of Convertible Preferred Stock and Stockholders’ Deficit
For
the Years ended December 31, 2022 and 2021
| | |
Series
A Convertible | | |
Series
A-1 Convertible | | |
Series
B Convertible | | |
| | |
Additional | | |
| | |
Total | |
| | |
Preferred
Stock | | |
Preferred
Stock | | |
Preferred
Stock | | |
Common
Stock | | |
Paid-In | | |
Accumulated | | |
Stockholders’ | |
| | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Capital | | |
Deficit | | |
Deficit | |
| Balance, December 31, 2020 | |
| 205,385 | | |
$ | 3,272,944 | | |
| 380,657 | | |
$ | 14,100,533 | | |
| 391,303 | | |
$ | 16,321,065 | | |
| 1,025,353 | | |
$ | 103 | | |
$ | 553,534 | | |
$ | (17,694,511 | ) | |
$ | (17,140,774 | ) |
| Stock-based compensation | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| 306,055 | | |
| — | | |
| 306,055 | |
| Exercise of stock options | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| 17,750 | | |
| 1 | | |
| 8,574 | | |
| — | | |
| 8,475 | |
| Net
loss | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| - | | |
| — | | |
| — | | |
| (8,939,675 | ) | |
| (8,939,675 | ) |
| Balance, December 31, 2021 | |
| 205,385 | | |
| 3,272,944 | | |
| 380,657 | | |
| 14,100,533 | | |
| 391,303 | | |
| 16,321,065 | | |
| 1,043,103 | | |
| 104 | | |
| 868,163 | | |
| (26,634,186 | ) | |
| (25,765,919 | ) |
| Stock-based
compensation | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| 184,465 | | |
| — | | |
| 184,465 | |
| Exercise of stock options | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| 888 | | |
| — | | |
| 1,510 | | |
| — | | |
| 1,510 | |
| Net
loss | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| (10,680,366 | ) | |
| (10,680,366 | ) |
| Balance,
December 31, 2022 | |
| 205,385 | | |
$ | 3,272,944 | | |
| 380,657 | | |
$ | 14,100,533 | | |
| 391,303 | | |
$ | 16,321,065 | | |
| 1,043,991 | | |
$ | 104 | | |
$ | 1,054,138 | | |
$ | (37,314,552 | ) | |
$ | (36,260,310 | ) |
See
accompanying notes
AZITRA,
INC.
Statements
of Cash Flows
| | |
Years
Ended December 31, | |
| | |
2022 | | |
2021 | |
| Cash flows
from operating activities: | |
| | | |
| | |
| Net
loss | |
$ | (10,680,366 | ) | |
$ | (8,939,675 | ) |
| Adjustments
to reconcile net loss to net cash and cash equivalents used by operating activities: | |
| | | |
| | |
| Depreciation
and amortization | |
| 125,885 | | |
| 84,772 | |
| Amortization
of debt discount | |
| 7,981 | | |
| 7,632 | |
| Amortization
of right-of-use assets | |
| 280,092 | | |
| - | |
| Accrued
interest on convertible notes | |
| 164,611 | | |
| 59,181 | |
| Stock-based
compensation | |
| 184,465 | | |
| 306,055 | |
| Change
in fair value of warrant liability | |
| (821 | ) | |
| (989 | ) |
| Change
in fair value of convertible notes | |
| 1,250,000 | | |
| - | |
| Forgiveness
of Payroll Protection Program loan | |
| - | | |
| (230,685 | ) |
| Loss on
disposal of property and equipment | |
| 7,923 | | |
| - | |
| Changes
in operating assets and liabilities: | |
| | | |
| | |
| Accounts
receivable | |
| (21,953 | ) | |
| (37,573 | ) |
| Prepaid
expenses | |
| (49,844 | ) | |
| 163,952 | |
| Other
assets | |
| 694 | | |
| (919 | ) |
| Tax credits
receivable | |
| 103,645 | | |
| 231,745 | |
| Income
tax receivable | |
| (15,024 | ) | |
| - | |
| Accounts
payable and accrued expenses | |
| 420,752 | | |
| 282,976 | |
| Operating
lease liability | |
| (268,509 | ) | |
| - | |
| Income
tax payable | |
| - | | |
| (8,831 | ) |
| Contract
liabilities | |
| 141,000 | | |
| 15,000 | |
| Net cash and cash equivalents
used by operating activities | |
| (8,349,469 | ) | |
| (8,067,359 | ) |
| | |
| | | |
| | |
| Cash flows
from investing activities: | |
| | | |
| | |
| Purchases
of property and equipment | |
| (33,101 | ) | |
| (446,136 | ) |
| Proceeds
from sale of property and equipment | |
| 4,250 | | |
| - | |
| Capitalization
of deferred patent costs | |
| (180,802 | ) | |
| (196,267 | ) |
| Capitalization of license | |
| (65,510 | ) | |
| - | |
| Capitalization
of patent and trademark costs | |
| (61,598 | ) | |
| (9,872 | ) |
| Net cash and cash equivalents
used by investing activities | |
| (336,761 | ) | |
| (652,275 | ) |
| | |
| | | |
| | |
| Cash flows
from financing activities: | |
| | | |
| | |
| Proceeds
from convertible notes, net of issuance costs | |
| 4,350,000 | | |
| 984,387 | |
| Payment
of deferred offering costs | |
| (216,886 | ) | |
| - | |
| Proceeds
from exercise of stock options | |
| 1,510 | | |
| 8,475 | |
| Net
cash and cash equivalents provided by financing activities | |
| 4,134,624 | | |
| 992,862 | |
| | |
| | | |
| | |
| Net decrease in cash and cash
equivalents | |
| (4,551,606 | ) | |
| (7,726,772 | ) |
| Cash
and cash equivalents at the beginning of the year | |
| 8,044,262 | | |
| 15,771,034 | |
| Cash
and cash equivalents at the end of the year | |
$ | 3,492,656 | | |
$ | 8,044,262 | |
See
accompanying notes
AZITRA,
INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
For The Years Ended December 31, 2022 and 2021
1.
Organization and Nature of Operations
Azitra,
Inc. was founded on January 2, 2014. It is a synthetic biology company focused on screening and genetically engineering microbes of the
skin. The mission is to discover and develop novel therapeutics to create a new paradigm for treating skin disease. The Company’s
discovery platform is screened for naturally occurring bacterial cells with beneficial effects. These microbes are then genomically sequenced
and engineered to make cellular therapies, recombinant therapeutic proteins, peptides and small molecules for precision treatment of
dermatology diseases.
The
Company maintains a location in Montreal, Canada for certain research activities. This location and operations completed there remained
consistent throughout 2021 and 2022. The Company also opened a manufacturing and laboratory space in Groton, Connecticut during 2021.
Going
Concern Matters
The
financial statements have been prepared on the going concern basis, which assumes that the Company will continue in operation for the
foreseeable future and which contemplates the realization of assets and liquidation of liabilities in the normal course of business.
However, management has identified the following conditions and events that created an uncertainty about the ability of the Company to
continue as a going concern. As of and for the year ended December 31, 2022, the Company has an accumulated deficit of $37.3 million,
a loss from operations of $9.5 million and used $8.6 million to fund operations. These factors among others raise substantial doubt about
the Company’s ability to continue as a going concern.
Management
plans to continue to raise funds through equity and debt financing to fund operating and working capital needs, however, the Company
will require a significant amount of additional funds to complete the development of its product and to fund additional losses which
the Company expects to incur over the next few years. The Company is in an early clinical phase and therefore does not yet have product
revenue. In 2022, the Company issued convertible notes resulting in net cash proceeds of $4.4 million (see Note 7). There can be no assurance
that the Company will be successful in securing additional financing, if needed, to meet its operating needs.
These
conditions and events create an uncertainty about the ability of the Company to continue as a going concern through February 20, 2024
(one year after the date that the financial statements are available to be issued). The financial statements do not include any adjustments
that might be necessary should the Company be unable to continue as a going concern.
2.
Summary of Significant Accounting Policies
Basis
of Accounting
The
financial statements of the Company are prepared in accordance with United States generally accepted accounting principles.
Use
of Estimates
The
preparation of the financial statement in conformity with accounting principles generally accepted in the United States of America requires
management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the balance sheet.
While management believes the estimates and assumptions used in the preparation of the financial statement are appropriate, actual results
could differ from those estimates.
Cash
and Cash Equivalents
For
purposes of the balance sheets and statements of cash flows, the Company considers all cash on hand, demand deposits and all highly liquid
investments with original maturities of three months or less to be cash equivalents.
AZITRA,
INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
For The Years Ended December 31, 2022 and 2021
Accounts
Receivable
The
Company carries its accounts receivable at cost less an allowance for doubtful accounts. On a periodic basis, the Company evaluates its
accounts receivable and establishes an allowance for doubtful accounts based on a history of past write-offs, collections and current
conditions. There was no allowance for doubtful accounts at December 31, 2022 or 2021. Accounts receivable are written off when deemed
uncollectible. Recoveries of accounts receivable previously written off are recorded when received.
Deferred
offering costs
The
Company capitalizes deferred offering costs, which primarily consist of direct, incremental legal, professional, accounting, and other
third-party fees relating to the Company’s initial public offering. The deferred offering costs will be offset against IPO proceeds
upon the consummation of an offering. Should the planned IPO prove to be unsuccessful, these deferred costs, as well as additional expenses
to be incurred, will be charged to operations.
Property
and Equipment
Property
and equipment are recorded at cost. Depreciation is computed using the straight-line method over the estimated useful lives, which range
from 3 to 10 years. Expenditures for maintenance and repairs, which do not extend the economic useful life of the related assets, are
charged to operations as incurred. Gains or losses on disposal of property and equipment are reflected in the statements of operations
in the period of disposal.
Right
of Use Assets
In
February 2016, the Financial Accounting Standards Board (“FASB”) issued ASU No. 2016-02, Leases (“Topic 842”).
ASU 2016-02 requires lessees to present right-of-use (“ROU”) assets and lease liabilities on the balance sheet for all leases
with terms longer than 12 months. See Note 2 – Recently Adopted Accounting Pronouncements.
In
calculating the effect of ASU 2016-02, the Company elected the transition method thereby not restating comparable periods. The Company
elected to account for non-lease components as part of the lease component to which they relate. Lease accounting involves significant
judgments, including making estimates related to the lease term, lease payments, and discount rate. In accordance with the guidance,
the Company recognized ROU assets and lease liabilities for all leases with a term greater than 12 months.
The
Company has operating leases for buildings. Currently, the Company has 3 operating leases with a ROU asset and lease liability totaling
$1,418,502 as of January 1, 2022. The basis, terms and conditions of the leases are determined by the individual agreements. The Company’s
option to extend certain leases ranges from 36 – 52 months. All options to extend have been included in the calculation of the
ROU asset and lease liability. The leases do not contain residual value guarantees, restrictions, or covenants that could incur additional
financial obligations to the Company. There are no subleases, sale-leaseback, or related party transactions.
At
December 31, 2022, the Company had operating right-of-use assets with a net value of $1,116,697 and current and long-term operating lease
liabilities of $287,384 and $840,896, respectively.
Intangible
Assets
Intangible
assets consist of trademarks and patents. All costs directly related to the filing and prosecution of patent and trademark applications
are capitalized. Patents are amortized over their respective remaining useful lives upon formal approval. Trademarks have an indefinite
life.
The
Company accounts for other indefinite life intangible assets in accordance ASC Topic 350, Goodwill and Other Intangible Assets
(ASC 350). ASC 350 requires that intangible assets that have indefinite lives are required to be tested at least annually for impairment
or whenever events or changes in circumstances indicate that the carrying amount may not be recoverable. Intangible assets that have
finite lives will continue to be amortized over their useful lives. No impairment losses relating to intangible assets were recorded
in 2022 or 2021.
AZITRA,
INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
For The Years Ended December 31, 2022 and 2021
Deferred
Patent Costs
Deferred
patent costs represent legal and filing expenses incurred related to the submission of patent applications for patents pending approval.
These deferred costs will begin to be amortized over their estimated useful lives upon the formal approval of the patent. If the patent
is not approved, the costs associated with the patent will be expensed in the year the patent was rejected. No impairment losses relating
to deferred patent costs were recorded in the years ended December 31, 2022 and 2021.
Impairment
of Long-Lived Assets
In
accordance with ASC Topic 360-10, Accounting for the Impairment or Disposal of Long-Lived Assets (ASC 360-10), the Company’s
policy is to review its long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount
of an asset may not be recoverable. In connection with this review, the Company also reevaluates the periods of depreciation for these
assets. The Company recognizes an impairment loss when the sum of the undiscounted expected future cash flows from the use and eventual
disposition of the asset is less than its carrying amount. If an asset is considered to be impaired, the impairment to be recognized
is measured by the amount by which the carrying amount of the asset exceeds the fair value of the asset, which is determined using the
present value of the net future operating cash flows generated by the asset.
Convertible
Debt and Warrant Accounting
Warrants
The
Company accounts for warrants as either equity-classified or liability-classified instruments based on an assessment of the warrant’s
specific terms and applicable authoritative guidance in ASC 480, Distinguishing Liabilities from Equity (“ASC 480”)
and ASC 815, Derivatives and Hedging (“ASC 815”). The assessment considers whether the warrants are freestanding financial
instruments pursuant to ASC 480, meet the definition of a liability pursuant to ASC 480, and whether the warrants meet all of the requirements
for equity classification under ASC 815, including whether the warrants are indexed to the Company’s own common stock, among other
conditions for equity classification. This assessment, which requires the use of professional judgment, is conducted at the time of warrant
issuance and as of each subsequent quarterly period end date while the warrants are outstanding.
For
issued warrants that meet all of the criteria for equity classification, the warrants are required to be recorded as a component of additional
paid-in capital at the time of issuance. For issued warrants that do not meet all the criteria for equity classification, the warrants
are required to be recorded at their initial fair value on the date of issuance, and each balance sheet date thereafter. Changes in the
estimated fair value of the warrants are recognized as a non-cash gain or loss on the statements of operations under Other Income/loss.
Convertible
Debt
When
the Company issues debt with a conversion feature, it first assesses whether the conversion feature meets the requirements to be accounted
for as stock settled debt. If it does not meet those requirements then it is assessed on whether the conversion feature should be bifurcated
and treated as a derivative liability, as follows: a) one or more underlyings, typically the price of our common stock; b) one or more
notional amounts or payment provisions or both, generally the number of shares upon conversion; c) no initial net investment, which typically
excludes the amount borrowed; and d) net settlement provisions, which in the case of convertible debt generally means the stock received
upon conversion can be readily sold for cash. An embedded equity-linked component that meets the definition of a derivative does not
have to be separated from the host instrument if the component qualifies for the scope exception for certain contracts involving an issuer’s
own equity. The scope exception applies if the contract is both a) indexed to its own stock; and b) classified in stockholders’
equity in its statement of financial position.
AZITRA,
INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
For The Years Ended December 31, 2022 and 2021
Convertible
Preferred Stock
As
the Convertible Preferred stockholders have liquidation rights in the event of a deemed liquidation event that, in certain situations,
are not solely within the control of the Company and would require the redemption of the then-outstanding Convertible Preferred Stock,
the Company classifies the Convertible Preferred Stock in mezzanine equity on the balance sheet. Due to the fact that the occurrence
of a deemed liquidation event is not currently probable, the carrying value of the Convertible Preferred Stock is not being accreted
to its redemption value. Subsequent adjustments to the carrying value of the Convertible Preferred Stock would be made only when a deemed
liquidation event becomes probable.
Revenue
The
Company follows the five steps to recognize revenue from contracts with customers under ASC 606, Revenue from Contracts with Customers
(“ASC 606”), which are:
| ● | Step
1: Identify the contract(s) with a customer |
| ● | Step
2: Identify the performance obligations in the contract |
| ● | Step
3: Determine the transaction price |
| ● | Step
4: Allocate the transaction price to the performance obligations in the contract |
| ● | Step
5: Recognize revenue when (or as) a performance obligation is satisfied |
The
Company generates service revenue through a joint development agreement with a research partner. The Company recognizes revenue related
to the research and development aspects of the agreement over time using the input method as work is performed on the contract.
The
Company also generates grant revenue, which represents monies received on contracts with various federal agencies and nonprofit research
institutions for general research conducted by the Company to further their product development and are therefore considered contributions
to the Company. The contracts are generally for periods of one year or more and can be cancelled by either party. The Company concluded
that the grant arrangements do not meet the criteria to be treated as a collaborative arrangement under FASB ASC Topic 808 as the Company
is the only active participant in the arrangement. The grant arrangements also do not meet the criteria for revenue recognition under
Topic 606, as the U.S. Government would not meet the definition of a customer.
Amounts
earned under these grant contracts are recorded as a negative research & development expense when eligible expenses are incurred
and the right to payment is realizable or realized and earn The Company believes this policy is consistent with Topic 606, to ensure
that recognition reflects the transfer of promised goods or services to customers in an amount that reflects the consideration that the
Company expects to be entitled to in exchange for those goods or services, even though there is no exchange as defined in Topic 606.
Additionally, the Company has determined that the recognition of amounts received as costs are incurred and amounts become realizable
is analogous to the concept of transfer of control of a service over time under Topic 606.
Receipts
of grant awards in advance, which are payable back to the funding agency if not used in accordance with conditions in the grants related
to allowable costs or receipt of funding from research partners related to service revenue arrangements before work is performed on the
contract are classified as contract liabilities in the accompanying balance sheets.
Research
and Development
The
Company accounts for research and development costs in accordance with Accounting Standards Codification (ASC) subtopic 730-10, Research
and Development. Accordingly, internal research and development costs are expensed as incurred. Research and development costs consist
of costs related to labor, materials and supplies. Research and development costs incurred were $5,920,538 and $5,380,102 during the
years ended 2022 and 2021, respectively.
AZITRA,
INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
For The Years Ended December 31, 2022 and 2021
At
December 31, 2022 and 2021, the Company has a state tax credit receivable of $32,459 and $71,396, respectively, for pending refunds related
to the selling of research and development tax credits back to the State of Connecticut. At December 31, 2022 and 2021, the Company has
$28,925 and $89,855, respectively for pending refunds related to Canadian Scientific Research and Experimental Development credits. At
December 31, 2022 and 2021, the Company has also recorded $8,282 and $12,060, respectively, related to refunds of Canadian Goods and
Services Tax (GST) and Quebec Sales Tax (QST). Receipts of refunds are recorded in other income on the statements of operations.
Stock-Based
Compensation
The
Company accounts for stock-based compensation in accordance with ASC 718, Compensation-Stock Compensation (ASC 718). ASC 718 requires
employee stock options and rights to purchase shares under stock participation plans to be accounted for at fair value. ASC 718 requires
that compensation costs related to share-based payment transactions be recognized as operating expenses in the financial statements.
Under this method, compensation costs for all awards granted or modified are measured at estimated fair value at date of grant and are
included as compensation expense over the vesting period during which an employee provides service in exchange for the award. For awards
with a performance condition that affects vesting, the Company recognizes compensation expense when it is determined probable that the
performance condition will be achieved.
The
Company uses a Black-Scholes option pricing model to determine fair value of its stock options. The Black-Scholes model includes various
assumptions, including the value of the underlying common stock, the expected life of stock options, the expected volatility and the
expected risk-free interest rate. These assumptions reflect the Company’s best estimates, but they involve inherent uncertainties
based on market conditions generally outside of the control of the Company. As a result, if other assumptions had been used, stock-based
compensation cost could have been materially impacted. Furthermore, if the Company uses different assumptions for future grants, stock-based
compensation cost could be materially impacted in future periods.
The
Company accounts for equity instruments issued to non-employees in accordance with the provisions of ASC 718 as updated by Accounting
Standards Update (ASU) No. 2018-07, Improvements to Nonemployee Share-Based Payment Accounting, which expands the scope of ASC
718 to include share based payment transactions to non-employees.
The
following assumptions are used in valuing options issued using the Black-Scholes option pricing model:
Expected
Volatility. The expected volatility of the Company’s shares is estimated based on the Company’s external valuation.
Expected
Term. The expected term of options is estimated using the simplified method which is based on the vesting period and contractual
term for each grant, or for each vesting-tranche for awards with graded vesting.
Underlying
Common Stock Value. The underlying common stock value of the Company’s shares is estimated by a third party valuation expert.
Risk-free
Interest Rate. The Company bases the risk-free interest rate on the implied yield available on a U.S. Treasury note with terms equal
to the expected term of the underlying grant.
Dividend
Yield. The Black-Scholes valuation model calls for a single expected dividend yield as an input. The Company has not paid dividends
on Common stock in the past nor does it expect to pay dividends on Common stock in the near future. As such, the Company uses a dividend
yield percentage of zero.
AZITRA,
INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
For The Years Ended December 31, 2022 and 2021
Income
Taxes
The
Company uses the liability method of accounting for income taxes, as set forth in ASC 740, Accounting for Income Taxes. Under
this method, deferred tax assets and liabilities are recognized for the expected future tax consequence of temporary differences between
the carrying amounts and the tax basis of assets and liabilities and net operating loss carry forwards, all calculated using presently
enacted tax rates (see Note 13).
Management
has evaluated the effect of ASC guidance related to uncertain income tax positions and concluded that the Company has no significant
financial statement exposure to uncertain income tax positions at December 31, 2022 and 2021. The Company’s income tax returns
have not been examined by tax authorities through December 31, 2022. The Company is not currently under audit by any tax jurisdiction.
Fair
Value Measurements
The
Company carries certain liabilities at fair value on a recurring basis. A fair value hierarchy that consists of three levels is used
to prioritize the inputs to fair value valuation techniques:
| ● | Level
1 – Inputs are based upon observable or quoted prices for identical instruments traded
in active markets. |
| ● | Level
2 – Inputs are based upon quoted prices for similar instruments in active markets,
quoted prices for identical or similar instruments in markets that are not active, and model-based
valuation techniques for which all significant assumptions are observable in the market or
can be corroborated by observable market data for substantially the full term of the assets
or liabilities. |
| ● | Level
3 – Inputs are generally unobservable and typically reflect management’s estimates
of assumptions that market participants would use in pricing the asset or liability. The
fair values are therefore determined using model-based techniques that include option pricing
models, discounted cash flow models, and similar techniques. |
In
determining fair value, the Company utilizes valuation techniques that maximize the use of observable inputs and minimize the use of
unobservable inputs to the extent possible as well as considers counterparty credit risk in its assessment of fair value.
Recently
Adopted Accounting Pronouncements
In
February 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) 2016-02,
Leases (Topic 842). This ASU requires a lessee to recognize a right-of-use asset and a lease liability under most operating leases in
its balance sheet. The ASU is effective for annual and interim periods beginning after December 15, 2021. The Company adopted ASU 2016-02
on January 1, 2022. See Note 15 – Operating Leases.
In
December 2019, the FASB issued ASU No. 2019-12, Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes. This standard
simplifies the accounting for income taxes through the removal of various exceptions previously provided, as well as providing additional
reporting requirements for income taxes. The ASU is effective for the Company on January 1, 2022. The Company has adopted this standard
effective January 1, 2022, which did not have a material impact to the financial statements.
In
August 2020, the FASB issued ASU No. 2020-06, Debt – Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives
and Hedging – Contracts in Entity’s Own Equity (Subtopic 815-40), which simplifies the accounting for certain financial
instruments with characteristics of liabilities and equity, including convertible instruments and contracts on an entity’s own
equity. This standard will be effective for the Company on January 1, 2024, with early adoption permitted (but no earlier than fiscal
years beginning after December 15, 2020). The Company has adopted this standard effective January 1, 2021, which did not have a material
impact to the financial statements.
Management
does not believe that any other recently issued, but not yet effective, accounting standards could have a material effect on the accompanying
financial statements. As new accounting pronouncements are issued, the Company will adopt those that are applicable under the circumstances.
AZITRA,
INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
For The Years Ended December 31, 2022 and 2021
Financial
Instruments
The
Company’s financial instruments are primarily comprised of accounts receivable, accounts payable, accrued liabilities, and long-term
debt. For accounts receivable, accounts payable and accrued liabilities, the carrying amount approximates fair value due to the short
term maturities of such instruments. The estimated fair value of the Company’s long-term debt approximates carrying value.
3.
Employee Retention Credit
The
CARES Act provides an employee retention credit (“CARES Employee Retention credit”), which is a refundable tax credit against
certain employment taxes of up to $5,000 per employee for eligible employers. The tax credit is equal to 50% of qualified wages paid
to employees during a quarter, capped at $10,000 of qualified wages per employee through December 31, 2020. Additional relief provisions
were passed by the United States government, which extend and slightly expand the qualified wage caps on these credits through September
30, 2021. Based on these additional provisions, the tax credit is now equal to 70% of qualified wages paid to employees during a quarter,
and the limit on qualified wages per employee has been increased to $10,000 of qualified wages per quarter. In April 2022, the Company
determined it qualified for the tax credit under the CARES Act and recorded a receivable for $229,813 and recognized the amounts as other
income on the statement of operations. The Company received full payment for the amount in September 2022.
4.
Property and Equipment
Property
and equipment consisted of the following at December 31, 2022 and 2021:
| | |
2022 | | |
2021 | |
| Lab equipment | |
$ | 1,034,579 | | |
$ | 1,016,737 | |
| Computer equipment | |
| 30,825 | | |
| 30,825 | |
| Furniture and fixtures | |
| 24,316 | | |
| 24,316 | |
| Leasehold improvements | |
| 28,855 | | |
| 28,855 | |
| Building equipment | |
| 14,932 | | |
| 14,932 | |
| |
| 1,133,507 | | |
| 1,115,665 | |
| Less: accumulated depreciation | |
| (286,549 | ) | |
| (168,984 | ) |
| Net property and equipment | |
$ | 846,958 | | |
$ | 946,681 | |
Depreciation
expense was $120,651 and $81,866 for the years ended December 31, 2022 and 2021, respectively.
5.
Intangible Assets
Intangible
assets consisted of the following at December 31:
2022:
| | |
Estimated
Useful Life | |
Gross
Amount | | |
Accumulated
Amortization | | |
Net
Amount | |
| Trademarks | |
Indefinite | |
$ | 53,999 | | |
$ | - | | |
$ | 53,999 | |
| Patents | |
17 years | |
| 108,198 | | |
| 8,140 | | |
| 100,058 | |
| License
agreement | |
17
years | |
| 65,510 | | |
| - | | |
| 65,510 | |
| Intangible
assets | |
| |
$ | 227,707 | | |
$ | 8,140 | | |
$ | 219,567 | |
AZITRA,
INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
For The Years Ended December 31, 2022 and 2021
2021:
| | |
Estimated
Useful Life | |
Gross
Amount | | |
Accumulated
Amortization | | |
Net
Amount | |
| Trademarks | |
Indefinite | |
$ | 50,955 | | |
$ | - | | |
$ | 50,955 | |
| Patents | |
17
years | |
| 49,644 | | |
| 2,906 | | |
| 46,738 | |
| Intangible
assets | |
| |
$ | 100,599 | | |
$ | 2,906 | | |
$ | 97,693 | |
During
2022 and 2021, amortization expense related to intangible assets was $5,234 and $2,906, respectively.
Expected
amortization expense is as follows for the years ending December 31:
| 2023 | |
$ | 2,920 | |
| 2024 | |
| 2,920 | |
| 2025 | |
| 2,920 | |
| 2026 | |
| 2,920 | |
| 2027 | |
| 2,920 | |
| Thereafter | |
| 85,456 | |
| Total | |
$ | 100,058 | |
6.
Accrued Expenses
Accrued
expenses consisted of the following at December 31, 2022 and 2021:
| | |
2022 | | |
2021 | |
| Employee payroll
and bonuses | |
$ | 371,010 | | |
$ | 138,671 | |
| Vacation | |
| 27,082 | | |
| 43,473 | |
| Research and development projects | |
| 316,389 | | |
| 149,711 | |
| Interest | |
| 223,792 | | |
| 59,181 | |
| Professional fees | |
| 24,502 | | |
| 58,892 | |
| Other | |
| 31,186 | | |
| 25,557 | |
| | |
$ | 993,961 | | |
$ | 475,485 | |
The
Company accrues expenses related to development activities performed by third parties based on an evaluation of services received and
efforts expended pursuant to the terms of the contractual arrangements. Payments under some of these contracts depend on research and
non-clinical trial milestones. There may be instances in which payments made to the Company’s vendors will exceed the level of
services provided and result in a prepayment of expense. In accruing service fees, the Company estimates the period over which services
will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level
of effort varies from the estimate, the Company will adjust the accrual or prepaid expense accordingly. The Company has not experienced
any material differences between accrued costs and actual costs incurred since its inception.
AZITRA,
INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
For The Years Ended December 31, 2022 and 2021
7.
Convertible Debt
In
September 2022, the Company entered into a Convertible Note Purchase Agreement (the Agreement) to issue up to $4,500,000 convertible
promissory notes. On the same day, the Company entered into convertible promissory notes (2022 Convertible Notes) with three investors
totaling $4,350,000. The 2022 Convertible Notes mature on January 13, 2023 or the occurrence of an Event of Default (as defined) and
bear interest at a rate of 8% per annum which shall accrue but is not due and payable until conversion or full repayment of outstanding
principal. The principal and interest outstanding under the 2022 Convertible Notes is automatically converted a) upon the closing of
a Qualified Financing resulting in gross proceeds to the Company of at least $20 million into securities issued in connection with the
Qualified Financing, at a discount of 30% per share; b) upon the closing of a Change of Control event into shares of capital stock of
the Company or Series B preferred stock; and c) upon the closing of a Public Company Event, into shares of capital stock being issued
to investors equal to two-times (2x) the amount of the outstanding principal and accrued interest then outstanding divided by the public
offering price per share. The principal and interest outstanding under the 2022 Convertible Notes is convertible, at the option of the
holders, at the maturity date into a new class of Company’s Preferred Stock (Series C Preferred) equal to the quotient of the outstanding
principal amount plus interest divided by the Capped Price, which is defined as the price per share equal to the Valuation Cap of $30
million divided by the Company Capitalization, as defined in the Agreement.
The
Company accounts for the 2022 Convertible Notes under ASC 815. Under 815-15-25, the election can be at the inception of a financial instrument
to account for the instrument under the fair value option under ASC 825. The Company has made such election for the 2022 Convertible
Notes. Using the fair value option, the convertible promissory note is to be recorded at its initial fair value on the date of issuance,
and each balance sheet date thereafter. The Company evaluates the change based on the conversion price at the current market value. When
recognized, changes in the estimated fair value of the notes are recognized as a non-cash gain or loss in Other Income (Expense) on the
statements of operations. The Company recognized a change in fair value of the 2022 Convertible Notes of $1,250,000 for the year ended
December 31, 2022.
Effective
January 5, 2021, the Company entered into a Note Purchase Agreement to issue up to $2,000,000 of convertible promissory notes. On the
same date, the Company entered into a convertible promissory note (2021 Convertible Note) with one investor for $1,000,000. The 2021
Convertible Note bears interest at a rate of 6% per annum and is due and payable in full on January 5, 2023. The 2021 Convertible Note
automatically converts upon a qualified equity financing, as defined in the note agreement to the number of shares equal to all principal
and accrued interest divided by the conversion price of $48.00, which is subject to adjustment as defined in the note agreement. The
2021 Convertible Note is also optionally convertible as defined in the note agreement for certain non-qualified financing, a change in
control, or upon the maturity date of the 2021 Convertible Note. The Company incurred issuance costs of $15,613 related to the 2021 Convertible
Note, which has been recorded as a debt discount and were amortized over the term of the 2021 Convertible Note. The issuance costs were
fully amortized at December 31, 2022.
The
Company evaluated the terms and conditions of the Note Purchase Agreement related to the 2021 Convertible Note in order to assess the
accounting considerations under ASC 480 – Distinguishing Liabilities from Equity, and ASC 815 – Derivatives and Hedging.
The Company determined the Convertible Note does not meet any of the criteria to be accounted pursuant to an ASC 480 liability. The Company
also assessed the embedded features pursuant to the guidance in ASC 815 and determined the embedded features do not meet any of the criteria
for bifurcation.
Convertible
notes payable consisted of the following at December 31:
| | |
2022 | | |
2021 | |
| 2021 Convertible
Note | |
$ | 1,000,000 | | |
$ | 1,000,000 | |
| 2022
Convertible Notes | |
| 5,600,000 | | |
| — | |
| | |
| 6,600,000 | | |
| 1,000,000 | |
| Debt
issuance costs | |
| — | | |
| (7,981 | ) |
| | |
$ | 6,600,000 | | |
$ | 992,019 | |
There
was $7,981 and $7,632 amortized related to the debt issuance costs during the years ended December 31, 2022 and 2021, respectively. Interest
accrued on the convertible notes was $223,792 and $59,181 at December 31, 2022 and 2021, respectively.
AZITRA,
INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
For The Years Ended December 31, 2022 and 2021
8.
Payroll Protection Program Loan
On
April 10, 2020 the Company received a loan in the amount of $230,685 under the Payroll Protection Program (PPP Loan). The loan accrued
interest at a rate of 1% and had an original maturity date of two years which could be extended to five years by mutual agreement of
the Company and the lender.
Under
the requirements of the CARES Act, as amended by the PPP Flexibility Act and Consolidated Appropriations Act, 2021, proceeds could only
be used for the Company’s eligible payroll costs (with salary capped at $100,000 on an annualized basis for each employee), or
other eligible costs related to rent, mortgage interest utilities, covered operations expenditures, covered property damage, covered
supplier costs, and covered worker protection expenditures, in each case paid during the 24-week period following disbursement. The PPP
Loan could be fully forgiven if (i) proceeds are used to pay eligible payroll costs or other eligible costs and (ii) full-time employee
headcount and salaries are either maintained during the 24-week period following disbursement or restored by December 31, 2020.
The
Company received notification of full forgiveness of the PPP Loan on January 25, 2021 and has recorded the amount in other income on
the statement of operations.
9.
Stockholders’ Equity
Common
Stock
At
December 31, 2022 and 2021, respectively, per the Company’s amended and restated Certificate of Incorporation, the Company was
authorized to issue 100,000,000 shares of $0.0001 par value common stock. Refer to Note 19, Subsequent Events for more information related
to the Forward-Stock Split.
The
Company had 1,043,991 and 1,043,103 shares of common stock issued and outstanding as of December 31, 2022 and 2021, respectively.
Each
share of common stock entitles the holder to one vote on all matters submitted to a vote of the Company’s stockholders and the
holders of the Common Stock are entitled to elect one director of the Corporation.
The
Company currently has 8,504,678 shares of common stock reserved for future issuance for the potential conversion of the outstanding Preferred
Stock and the exercise of stock options and warrants outstanding at December 31, 2022.
Preferred
Stock
At
December 31, 2022 and 2021, respectively, per the Company’s amended and restated Certificate of Incorporation, the Company has
authorized 1,437,150 and 978,042 shares of $0.0001 par value preferred stock.
The
Series A, Series A-1, and Series B Preferred Stock have the following rights, preferences and privileges:
Conversion
The
preferred stock is convertible, at the option of the holder, into common shares based upon a predefined formula. A holder of preferred
stock may convert such shares into common shares at any time. For purpose of conversion, the initial conversion price is $2.2887 per
share (original issue price) for Series A Preferred Stock, $5.2817 per share (original issue price) for Series A-1 Preferred Stock, and
$6.1190 per share (original issue price) for Series B Preferred Stock, and is subject to adjustment as described in the Certificate of
Incorporation. Preferred stock will automatically convert into common shares upon the earlier of (a) an initial public offering with
gross proceeds in excess of $100,000,000 or (b) the date and time, or the occurrence of an event, specified by vote or written consent
of the required preferred stock shareholders, all outstanding Series A, Series A-1, and Series B Preferred Stock shall automatically
convert into common shares, at the then effective conversion rate.
AZITRA,
INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
For The Years Ended December 31, 2022 and 2021
Voting
Rights
The
holders of the Series A, Series A-1, and Series B Preferred Stock are entitled to vote on any matter presented to the stockholders of
the Corporation for their action or consideration at any meeting of stockholders of the Corporation (or by written consent of stockholders
in lieu of meeting), each holder of outstanding shares of preferred stock shall be entitled to cast the number of votes equal to the
number of whole shares of Common Stock into which the shares of preferred stock held by such holder are convertible as of the record
date for determining stockholders entitled to vote on such matter. The holders of the Series A and Series A-1 Preferred Stock are each
entitled to elect one director of the Corporation. The holders of the Series B Stock are entitled to elect two members of the Board.
Each class of preferred stock can remove from office such directors and to fill any vacancy caused by the resignation, death or removal
of such directors under certain circumstances as described in the Certificate of Incorporation.
Dividends
The
holders of Series A Preferred Stock are entitled to receive dividends at a rate of 8% per annum of the Series A original issue price
of $2.2887 per share on each outstanding share of Series A Preferred Stock (subject to appropriate adjustment in the event of any stock
dividend, stock split, combination or other similar recapitalization with respect to the Series A Preferred Stock). Dividends accumulate
from the original date of issuance of the Series A Preferred Stock, are cumulative and are payable upon declaration of the Board of Directors
or liquidation of the Company. At December 31, 2022, cumulative dividends on Series A Preferred Stock were $1,541,575.
The
holders of Series A-1 Stock are entitled to receive dividends at a rate of 8% per annum of the Series A-1 original issue price of $5.2817
per share on each outstanding share of Series A-1 (subject to appropriate adjustment in the event of any stock dividend, stock split,
combination or other similar recapitalization with respect to the Series A-1 Preferred Stock). Dividends are cumulative and are payable
upon declaration of the Board of Directors or liquidation of the Company. At December 31, 2022, cumulative dividends on Series A-1 Preferred
Stock were $4,332,153.
The
holders of Series B Stock are entitled to receive dividends at a rate of 8% per annum of the Series B original issue price of $6.1190
per share on each outstanding share of Series B (subject to appropriate adjustment in the event of any stock dividend, stock split, combination
or other similar recapitalization with respect to the Series B Preferred Stock). Dividends are cumulative and are payable upon declaration
of the Board of Directors or liquidation of the Company. At December 31, 2022, cumulative dividends on Series B Preferred Stock were
$3,135,585.
Liquidation
In
the event of any liquidation, dissolution or winding up of the Company, the holders of the preferred stock are entitled to receive, prior
to and in preference to the holders of the common shares, an amount equal to the Series A, Series A-1, or Series B Preferred Stock original
issue price, plus declared and/or accrued but unpaid dividends. In the event of any such liquidation event, after the payment of all
preferential amounts required to be paid to the holders of shares of preferred stock, the remaining assets of the Corporation available
for distribution to its stockholders shall be distributed among the holders of the shares of preferred stock and Common Stock, pro rata
based on the number of shares held by each such holder, treating for this purpose all such securities as if they had been converted into
Common Stock pursuant to the terms of the Certificate of Incorporation immediately prior to such liquidation event.
10.
Warrants
The
Company issued warrants to purchase 47,889 shares of common stock in 2018 in conjunction with convertible debt financing that have a
redemption provision providing the holder the right to have the Company redeem all or any portion of the warrant (or shares it has converted
into) at a purchase price equal to the fair market value of the shares as determined by the board of directors or an independent appraiser.
As a result of this redemption provision, the warrants have been classified as a liability in the financial statements based on ASC 480
– Distinguishing Liabilities from Equity. These warrants have an exercise price of $0.48 per share a term of 10 years. The warrants
are marked to market each reporting period. The fair value is $70,283 and $71,104 at December 31, 2022 and 2021, respectively. At December
31, 2022, the Company estimated the fair value of the warrants using the Black-Scholes option pricing model with the following assumptions:
Underlying common stock value of $1.70; Expected term of 5 years; Expected Volatility of 86.0%; Risk Free Interest Rate of 3.01%; and
Dividend Yield of 0%. At December 31, 2021, the Company estimated the fair value of the warrants using the Black-Scholes option pricing
model with the following assumptions: Underlying common stock value of $1.70; Expected term of 6 years; Expected Volatility of 86.0%;
Risk Free Interest Rate of 1.35%; and Dividend Yield of 0%.
AZITRA,
INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
For The Years Ended December 31, 2022 and 2021
The
Company also issued warrants in 2016 and 2019 which did not meet the criteria under ASC 480 to be classified as a liability, and instead
meet equity classification criteria.
The
following table summarizes information about warrants outstanding at December 31, 2022:
| | |
| | |
Warrants
Outstanding | | |
Warrants
Exercisable | |
| Year
Granted | |
Exercise
Price | | |
Number
of Warrants at 12/31/2022 | | |
Weighted
Average Remaining Contractual Life | | |
Weighted
Average Exercise Price | | |
Number
of Warrants at 12/31/2022 | | |
Weighted
Average Remaining Contractual Life | | |
Weighted
Average Exercise Price | |
| 2016 | |
$ | 0.01 | | |
| 11,467 | | |
| | | |
$ | 0.01 | | |
| 11,467 | | |
| | | |
$ | 0.01 | |
| 2018 | |
$ | 0.48 | | |
| 47,889 | | |
| 3.7
years | | |
$ | 0.48 | | |
| 47,889 | | |
| 3.7
years | | |
$ | 0.48 | |
| 2019 | |
$ | 5.28 | | |
| 215,854 | | |
| 5.3
years | | |
$ | 5.28 | | |
| 215,854 | | |
| 5.3
years | | |
$ | 5.28 | |
| | |
| | | |
| 275,210 | | |
| 3.1
years | | |
$ | 4.23 | | |
| 275,210 | | |
| 3.1
years | | |
$ | 4.23 | |
11.
Stock Options
During
2016, the Company established the Azitra, Inc. 2016 Stock Incentive Plan (the Plan) which provides for the granting of stock options
and restricted shares to the Company’s employees, officers, directors, advisors and consultants. There are 1,490,595 shares available
for granting under the Plan at December 31, 2022 and 2021. Options vest over varying time frames.
During
the year ended December 31, 2022 and 2021, the Company granted 0 and 495,580, respectively, stock options to acquire shares of common
stock. The options vest over varying time frames between two and four years, have a life of ten years and an exercise price of $1.70.
During the years ended December 31, 2022 and 2021, the Company recognized stock compensation expense of $184,465 and $306,055, respectively,
relating to the issuance of service-based stock options. At December 31, 2022, there was $427,784 of unamortized compensation expense
that will be amortized over the remaining vesting period. At December 31, 2022, there were 93,152 performance-based options outstanding
with a fair value of $109,551. During the year ended December 31, 2022, the Company did not recognize any compensation expense for performance-based
options. The Company determined the options qualified as plain vanilla under the provisions of SAB 107 and the simplified method was
used to estimate the expected option life.
To
determine the estimated fair value of the options granted during 2021, the Company used the Black-Scholes option pricing model with the
following assumptions: Underlying common stock value of $1.70; Expected term of 7 years; Expected Volatility of 86.0%; Risk Free Interest
Rate of 0.66% - 1.26%; and Dividend Yield of 0%.
AZITRA,
INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
For The Years Ended December 31, 2022 and 2021
The
following table summarizes information about options outstanding and exercisable at December 31, 2022:
| | | |
Warrants
Outstanding | | |
Warrants
Exercisable | |
| Exercise
Price | | |
Number
of Options at 12/31/2022 | | |
Weighted
Average Remaining Contractual Life | | |
Weighted
Average Exercise Price | | |
Number
of Options at 12/31/2022 | | |
Weighted
Average Remaining Contractual Life | | |
Weighted
Average Exercise Price | |
| $ | 0.48 | | |
| 246,136 | | |
| 3.0
years | | |
$ | 0.48 | | |
| 246,136 | | |
| 3.0
years | | |
$ | 0.48 | |
| $ | 0.93 | | |
| 231,680 | | |
| 3.0
years | | |
$ | 0.93 | | |
| 231,680 | | |
| 3.0
years | | |
$ | 0.93 | |
| $ | 1.70 | | |
| 812,502 | | |
| 8.3
years | | |
$ | 1.70 | | |
| 411,707 | | |
| 8.0
years | | |
$ | 1.70 | |
| | | | |
| 1,290,318 | | |
| | | |
| | | |
| 889,523 | | |
| | | |
| | |
Total
stock option activity for the years ended December 31, 2022 and 2021 is summarized as follows:
| | |
Shares | | |
Weighted
Average Exercise Price | |
| Outstanding at December 31, 2020 | |
| 959,827 | | |
$ | 1.11 | |
| Granted | |
| 495,580 | | |
| 1.70 | |
| Exercised | |
| (17,750 | | |
| 0.48 | |
| Forfeited | |
| (55,749 | | |
| 1.03 | |
| Outstanding at December 31, 2021 | |
| 1,381,908 | | |
$ | 1.34 | |
| Exercised | |
| (888 | | |
| 1.70 | |
| Forfeited | |
| (90,702 | | |
| 5.91.41 | |
| Outstanding at December
31, 2022 | |
| 1,290,318 | | |
$ | 1.33 | |
There
are 157,989 shares available for future grant under the Plan at December 31, 2022.
12.
Fair Value Measurements
The
following tables summarize the fair values and levels within the fair value hierarchy in which the fair value measurements fall for assets
and liabilities measured on a recurring basis as of:
December
31, 2022 Description | |
Level
1 | | |
Level
2 | | |
Level
3 | | |
Total | |
| Liabilities: | |
| | | |
| | | |
| | | |
| | |
| Common
stock warrants | |
| | | |
| | | |
$ | 70,283 | | |
$ | 70,283 | |
| 2022
Convertible Notes | |
| | | |
| | | |
| 5,600,000 | | |
| 5,600,000 | |
| Total | |
$ | — | | |
$ | — | | |
$ | 5,670,283 | | |
$ | 5,670,283 | |
December
31, 2021 Description | |
Level
1 | | |
Level
2 | | |
Level
3 | | |
Total | |
| Liabilities: | |
| | | |
| | | |
| | | |
| | |
| Common
stock warrants | |
$ | | | |
$ | | | |
$ | 71,104 | | |
$ | 71,104 | |
| Balance at January 1, 2021 | |
$ | 72,093 | |
| Change in fair value of warrants | |
| (989 | ) |
| Balance at December 31, 2021 | |
| 71,104 | |
| Change
in fair value of warrants | |
| (821 | ) |
| Issuance of 2022 Convertible
Notes | |
| 4,350,000 | |
| Change
in fair value of 2022 Convertible Notes | |
| 1,250,000 | |
| | |
$ | 5,670,283 | |
AZITRA,
INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
For The Years Ended December 31, 2022 and 2021
Fluctuation
in the fair value of the Company’s Common stock is the primary driver for the change in the Common Stock Warrant liability valuation
during each year. As the fair value of the Common stock increases the value to the holder of the instrument generally increases.
Fluctuations
in the various inputs, including the enterprise value, time to liquidity, volatility, and discount rate are the primary drivers for the
changes in valuation of the 2022 Convertible Notes each reporting period. As the fair value of the enterprise value, estimated time to
liquidity, volatility, and discount rate increase, the value to the holder of the 2022 Convertible Notes generally increases.
13.
Income Taxes
Deferred
income taxes are provided on temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes
and those for income tax reporting purposes. Deferred income tax assets / (liabilities) as of December 31, 2022 and 2021 are as follows:
| | |
2022 | | |
2021 | |
| Deferred tax assets: | |
| | | |
| | |
| Net
operating loss carryforwards | |
$ | 7,870,665 | | |
$ | 6,994,967 | |
| Tax credits | |
| 1,729,421 | | |
| 1,216,246 | |
| Depreciation
and amortization | |
| 1,189,975 | | |
| | |
| Accrued
expenses | |
| 81,876 | | |
| 48,288 | |
| Other | |
| 28,057 | | |
| 25,240 | |
| Total deferred tax assets | |
| 10,899,994 | | |
| 8,284,741 | |
| | |
| | | |
| | |
| Deferred tax liabilities: | |
| | | |
| | |
| Depreciation
and amortization | |
| | | |
| | |
| Total
deferred tax liabilities | |
| — | | |
| (234,616 | ) |
| Valuation allowance | |
| | | |
| (234,616 | ) |
| Net
deferred tax (liability) asset | |
| (10,899,994 | ) | |
| (8,050,125 | ) |
| | |
$ | — | | |
$ | — | |
The
Company has federal net operating loss carryforwards of approximately $29,192,000 and $25,954,000 for the tax years ending December 31,
2022 and 2021, respectively, of which $1,285,000 will expire in tax years 2036 through 2037 and approximately $27,907,000 which does
not expire. The Company has state net operating loss carryforwards of approximately $29,374,000 and $25,947,000 for the tax years ending
December 31, 2022 and 2021, respectively, which will expire in tax years 2036 through 2042.
The
Company has federal research tax credits of approximately $1,235,000 and $859,000 for the tax years ending December 31, 2022 and 2021,
respectively, which expire in tax years 2039 through 2042. The Company has state research tax credits of approximately $350,000 and $249,000
for the tax years ending December 31, 2022 and 2021, respectively, of which $60,000 will expire in tax year 2036, $100,000 will expire
in tax year 2037 and the remainder can be carried forward indefinitely. The Company has Canadian research tax credits of approximately
$218,000 and $138,000 for the tax years ending December 31, 2022 and 2021, respectively, which expire in tax years 2039 through 2042.
AZITRA,
INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
For The Years Ended December 31, 2022 and 2021
The
U.S. Internal Revenue Code Section 382 imposes an annual limit on the ability of a corporation that undergoes a greater than 50% ownership
change to use its net operating loss carry forwards to reduce its tax liability. If in the future the Company undergoes an ownership
change exceeding the 50% limitation threshold imposed by Section 382, the Company’s net operating loss carryforwards may be significantly
limited as to the amount of use in a particular year. In addition, all or a portion of the Company’s net operating loss carryforwards
incurred before 2018, may expire unutilized.
The
realization of deferred tax assets is dependent upon the Company’s ability to generate future taxable income during the periods
in which the temporary differences become deductible. Based on the Company’s recent earnings history and projected future U.S.
earnings, management believes that it is more likely than not that its federal and state deferred tax assets will not be fully realized
in the foreseeable future. As a result of this assessment, management believes that a full valuation allowance against its net federal
and state deferred tax assets is required.
The
Company applies the provisions of ASC 740-10 to account for uncertain tax positions. ASC 740-10 addresses the determination of whether
tax benefits claimed or expected to be claimed on a tax return should be recorded in the financial statements. Under ASC 740-10, the
Company may recognize the tax benefit from an uncertain tax position only if it is more likely that not that the tax position will be
sustained on examination by the taxing authorities, based on the technical merits of the position. The Company has determined that it
has no significant uncertain tax positions requiring recognition and measurement under ASC 740-10.
The
Company is subject to U.S. federal income tax, Connecticut state income tax and Canada branch tax. The Company has not been audited by
the IRS, state, or foreign tax authorities in connection with income taxes. The Company’s tax years remain open to examination
for all federal and state tax matters until its net operating loss carryforwards are utilized and the applicable statute of limitations
have expired.
The
Company will recognize interest and penalties related to unrecognized tax benefits, if applicable, as a component of income tax expense.
14.
Net Loss Per Share
Basic
and diluted net loss per share were calculated as follows:
The
numerator for basic and diluted net loss per share is as follows:
| | |
For
the Year Ended December 31, | |
| | |
2022 | | |
2021 | |
| Net loss | |
$ | 10,680,366 | | |
$ | 8,939,675 | |
| Dividends
on preferred stock | |
| 2,768,984 | | |
| 2,768,984 | |
| Net
loss attributable to common shareholders | |
$ | 13,449,350 | | |
$ | 11,708,659 | |
The
denominator is as follows:
| | |
For
the Year Ended December 31, | |
| | |
2022 | | |
2021 | |
| Weighted average
common stock outstanding, basic and diluted | |
| 1,043,990 | | |
| 1,034,299 | |
| $0.01 warrants | |
| 11,467 | | |
| 11,467 | |
| Total | |
| 1,055,457 | | |
| 1,045,766 | |
AZITRA,
INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
For The Years Ended December 31, 2022 and 2021
Net
loss per share, basic and diluted is as follows:
| | |
For
the Year Ended December 31, | |
| | |
2022 | | |
2021 | |
| Net loss per share,
basic and diluted | |
$ | (12.74 | ) | |
$ | (11.20 | ) |
The
following potential common stock equivalents, presented based on amounts outstanding at each period end, were excluded from the calculation
of diluted net loss per share for the periods indicated because including them would have had an anti-dilutive effect:
| | |
December
31, | |
| | |
2022 | | |
2021 | |
| Options to purchase
shares of common stock | |
| 1,290,318 | | |
| 1,381,908 | |
| Warrants
outstanding | |
| 263,743 | | |
| 263,743 | |
| | |
| 1,554,061 | | |
| 1,645,651 | |
15.
Commitments and Contingencies
Legal
The
Company is subject to legal proceedings or claims which arise in the ordinary course of its business. Although occasional adverse decisions
or settlements may occur, the Company believes that the final disposition of such matters should not have a material adverse effect on
its financial position, results of operations or liquidity.
License
Agreement
Effective
January 26, 2022, the Company entered into an Exclusive License Agreement (the Agreement) with an unrelated third party. Under the Agreement,
the Company is granted an exclusive license for certain patents and a non-exclusive license for certain know-how. The Agreement continues
until the later of the expiration of the last to expire licensed patent or ten years after the first commercial sale of the first licensed
therapeutic or non-therapeutic product. The Company may terminate the Agreement at any time by providing at least 30 days written notice
to the third party. The Agreement is also terminated upon breach of a material obligation under the agreement or bankruptcy. Upon any
termination of the agreement, neither party is relieved of obligations incurred prior to the termination.
During
the year ended December 31, 2022, the Company capitalized payments made under this license agreement in the amount of $65,510. These
capitalized costs will be amortized over the life of the licensed patents, once issued.
Operating
Leases
The
Company leases office and lab space in Branford, CT; Groton, CT; and Laval, Quebec. The Company’s leases expire at various dates
through May 31, 2027. Most leases are for a fixed term and for a fixed amount. The Company is not a party to any leases that have step
rent provisions, escalation clauses, capital improvement funding or payment increases based on any index or rate.
During
2020, the Company entered into a new lease agreement for the Company’s primary office and laboratory space in Branford, CT. The
Branford lease requires monthly payments of $13,033 for the first year of the lease, which increases approximately 2% in each of the
following years. The Branford lease also requires the Company to pay a pro-rata share of common area maintenance.
AZITRA,
INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
For The Years Ended December 31, 2022 and 2021
During
May 2021, the Company entered into a new lease for office and laboratory space in Groton, CT. The Groton lease required monthly payments
of $4,234, which was increased to $6,824 in September 2021 upon leasing additional space. The Groton lease is initially for a one-year
term, with up to three additional years renewal available.
Future
minimum payments under non-cancelable operating leases with initial or remaining terms in excess of one year during each of the next
five years follow:
| 2023 | |
$ | 329,937 | |
| 2024 | |
| 335,988 | |
| 2025 | |
| 277,209 | |
| 2026 | |
| 204,159 | |
| 2027 | |
| 76,324 | |
| Total future undiscounted
lease payments | |
| 1,223,617 | |
| Less:
interest | |
| (95,337 | ) |
| Present
value of lease liabilities | |
$ | 1,128,280 | |
Rent
expense for all operating leases was $338,864 for the year ended December 31, 2022. The weighted average lease term for all operating
leases is 3.8 years. The weighted average discount rate for all operating leases is 4.25%.
16.
Retirement Plan
Effective
January 1, 2019, the Company sponsors a 401(k) plan that covers substantially all employees. In order to be eligible to participate,
an employee must complete two consecutive months of service and work a minimum of two hundred and fifty hours or work 1,000 hours in
their first year of service. Employees may make pre-tax deferrals upon meeting the Plan eligibility requirements. Effective January 1,
2020, the Plan was transitioned to a safe harbor plan in which highly compensated employees are not eligible for matching contributions
and non-highly compensated employees earn 100% match on first 3% contributed and 50% on the next 2% contributed. Total employer matching
contributions were $23,466 and $31,548 for the years ended December 31, 2022 and 2021, respectively.
17.
Concentration of Credit Risk
Financial
instruments that potentially subject the Company to credit risk consist principally of cash and accounts receivable.
For
the years ended December 31, 2022 and 2021, all grant revenue was from one grantor and three grantors, respectively, and all service
revenue was from one customer for both years.
The
cash balance identified in the balance sheet is held in an account with a financial institution and insured by the Federal Deposit Insurance
Corporation (FDIC) up to $250,000. At times, cash maintained on deposit may be in excess of FDIC limits.
In
early March 2020, there was a global outbreak of COVID-19 that has resulted in significant changes in the global economy. While the Company
has not experienced any disruptions to its business operations to date, these changes, including a potential economic downturn, and any
potential resulting direct or indirect negative impact to the Company cannot be determined, however they could have a prospective material
impact to the Company’s business, cash flows and liquidity.
AZITRA,
INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
For The Years Ended December 31, 2022 and 2021
18.
Related Parties
The
Company earned service revenue from an entity who was also an investor in the Company’s Series B Preferred Stock financing (see
Note 9). Total related party revenue was $284,000 and $110,000 for the years ended December 31, 2022 and 2021, respectively and accounts
receivable due from the related party was $175,000 and $125,000 at December 31, 2022 and 2021, respectively. Contract liabilities from
the related party were $156,000 and $15,000 at December 31, 2022 and 2021, respectively.
In
September 2022 the Company entered into a convertible promissory note totaling $4,350,000 of which $4,000,000 was attributable to an
entity who was also an investor in the Company’s Series A, A-1, and B Preferred Stock financing. (See Note 7)
19.
Subsequent Events
The
Company has evaluated events occurring between December 31, 2022 and June 13, 2023, the date the financial statements were available
to be issued.
In
January 2023, the Company elected to convert the 2021 Convertible Note (see Note 7), including interest accrued but not yet paid of $120,058
at a conversion price of $48.00 into 23,335 shares of its Series B Preferred Stock in accordance with the terms outlined in the Note
Purchase Agreement.
In
February 2023, the 2022 Convertible Notes were amended to extend the maturity date to March 31, 2023 and to change the conversion price
upon a Qualified Financing or Change in Control event to $30 million divided by the number of shares of the Company’s common stock
issued and outstanding, on a fully diluted basis, immediately prior to the close of the Qualified Financing or Change in Control event.
In
March 2023, the Company’s Board of Directors and stockholders approved the 2023 Stock Incentive Plan (“2023 Plan”);
however, no grants shall be made until the date which the proposed forward stock split and increase in the authorized shares of common
stock becomes effective which is expected to occur upon the effectiveness of our registration statement of which this prospectus forms
a part. The 2023 Plan allows the Committee to grant up to 2,000,000 shares of Common Stock in the form of incentive and non-statutory
stock options, restricted stock awards, restricted stock units, and other stock-based awards to employees, directors, and non-employees.
As of March 20, 2023, there were no awards issued under the 2023 Plan.
On
May 17, 2023, the Board of Directors approved a forward stock split at a ratio of 7.1-for-1 upon the effectiveness of the registration
statement of which this prospectus forms a part. The Board of Directors also approved an increase in the authorized shares of the Company’s
Common Stock to 100,000,000 along with a change in the Common Stock par value to $0.0001. All references to common stock, options to
purchase common stock, share data, per share data and related information contained in the consolidated financial statements and related
footnotes have been retrospectively adjusted, where applicable, to reflect the effect of the stock split, change in par value of our
Common Stock, and the adjustment of the preferred stock conversion ratios, where applicable. Accordingly, an adjustment was made between
common stock and additional paid-in-capital in the consolidated balance sheets to reflect the new values after the stock split.
AZITRA,
INC.
Balance
Sheets
Unaudited
| | |
September
30, 2023 | | |
December
31, 2022 | |
| ASSETS | |
| (Unaudited) | | |
| | |
| Current assets: | |
| | | |
| | |
| Cash and cash
equivalents | |
$ | 4,400,327 | | |
$ | 3,492,656 | |
| Accounts receivable | |
| - | | |
| 182,820 | |
| Tax credits receivable | |
| 40,525 | | |
| 69,666 | |
| Income tax receivable | |
| 13,722 | | |
| 13,722 | |
| Deferred offering costs | |
| - | | |
| 216,886 | |
| Prepaid
expenses | |
| 409,170 | | |
| 160,133 | |
| Total current assets | |
| 4,863,744 | | |
| 4,135,883 | |
| | |
| | | |
| | |
| Property and equipment, net | |
| 736,423 | | |
| 846,958 | |
| | |
| | | |
| | |
| Other assets | |
| | | |
| | |
| Other assets | |
| 47,546 | | |
| 47,507 | |
| Financing lease right-of-use
asset | |
| 43,872 | | |
| - | |
| Operating lease right-of-use
asset | |
| 899,619 | | |
| 1,116,697 | |
| Intangible assets, net | |
| 162,240 | | |
| 219,567 | |
| Deferred
patent costs | |
| 736,800 | | |
| 800,831 | |
| Total other assets | |
$ | 1,890,077 | | |
$ | 2,184,602 | |
| | |
| | | |
| | |
| Total
assets | |
$ | 7,490,244 | | |
$ | 7,167,443 | |
| | |
| | | |
| | |
| LIABILITIES, PREFERRED STOCK,
AND STOCKHOLDERS’ EQUITY | |
| | | |
| | |
| Current liabilities: | |
| | | |
| | |
| Accounts payable | |
| 417,928 | | |
| 784,687 | |
| Current financing lease
liability | |
| 14,254 | | |
| - | |
| Current operating lease
liability | |
| 301,423 | | |
| 287,384 | |
| Accrued expenses | |
| 720,095 | | |
| 993,961 | |
| Contract
liabilities | |
| - | | |
| 156,000 | |
| Total current liabilities | |
| 1,453,700 | | |
| 2,222,032 | |
| | |
| | | |
| | |
| Long-term financing lease
liability | |
| 29,952 | | |
| - | |
| Long-term operating lease
liability | |
| 613,572 | | |
| 840,896 | |
| Warrant liability | |
| 60,933 | | |
| 70,283 | |
| Convertible
notes payable, net | |
| - | | |
| 6,600,000 | |
| Total liabilities | |
| 2,158,157 | | |
| 9,733,211 | |
| | |
| | | |
| | |
| Commitments and contingencies
(Note 13) | |
| | | |
| | |
| | |
| | | |
| | |
| Preferred stock: | |
| | | |
| | |
| Series A convertible
preferred stock; $0.0001 par value; 205,385 shares authorized at September 30, 2023 and December 31, 2022; 0 and 205,385 shares issued
and outstanding at September 30, 2023 and December 31, 2022 respectively; liquidation value of $0 and $3,337,506 at September 30,
2023 and December 31, 2022 respectively | |
| | | |
| 3,272,944 | |
| Series A-1 convertible
preferred stock; $0.0001 par value; 380,657 shares authorized at September 30, 2023 and December 31, 2022; 0 and 380,657 shares issued
and outstanding at September 30, 2023 and December 31, 2022 respectively; liquidation value of $0 and $14,274,638 as of September
30, 2023 and December 31, 2022, respectively | |
| | | |
| 14,100,533 | |
| Series B convertible
preferred stock; $0.0001 par value; 851,108 shares authorized at September 30, 2023 and December 31, 2022; 0 and 391,303 shares issued
and outstanding at September 30, 3023 and December 31, 2022, respectively; liquidation value of $0 and $17,000,159 as of September
30, 2023 December 31, 2022 respectively | |
| | | |
| 16,321,065 | |
| | |
| | | |
| | |
| Stockholders’ equity
(deficit) | |
| | | |
| | |
| Common stock; $0.0001
par value, 100,000,000 shares authorized at September 30, 2023 and December 31, 2022, 12,097,643 and 1,043,988 shares issued and
outstanding at September 30, 2023 and December 31, 2022, respectively | |
| 1,210 | | |
| 104 | |
| Additional paid-in capital | |
| 51,475,425 | | |
| 1,054,138 | |
| Accumulated
deficit | |
| (46,144,548 | ) | |
| (37,314,552 | ) |
| Total
stockholders’ equity (deficit) | |
| 5,332,087 | | |
| (36,260,310 | ) |
| Total
liabilities, preferred stock, and stockholders’ equity (deficit) | |
$ | 7,490,244 | | |
$ | 7,167,443 | |
The
accompanying notes are an integral part of these condensed financial statements.
AZITRA,
INC.
Statements
of Operations
Unaudited
| | |
For the Nine
Months | | |
For the Nine
Months | |
| | |
September
30, 2023 | | |
September
30, 2022 | |
| Service
revenue - related party | |
$ | 596,000 | | |
$ | 253,500 | |
| Total revenue | |
| 596,000 | | |
| 253,500 | |
| | |
| | | |
| | |
| Operating expenses: | |
| | | |
| | |
| General and administrative | |
| 3,443,559 | | |
| 2,583,818 | |
| Research
and development | |
| 2,188,795 | | |
| 4,425,195 | |
| Total
operating expenses | |
| 5,632,354 | | |
| 7,009,013 | |
| | |
| | | |
| | |
| Loss from operations | |
| (5,036,354 | ) | |
| (6,755,513 | ) |
| | |
| | | |
| | |
| Other income (expense): | |
| | | |
| | |
| Interest income | |
| 1,184 | | |
| 4,056 | |
| Interest expense | |
| (166,729 | ) | |
| (66,781 | ) |
| Employee retention credit | |
| - | | |
| 229,813 | |
| Forgiveness of accounts
payable | |
| 56,285 | | |
| - | |
| Change in fair value of
convertible note | |
| (3,630,100 | | |
| - | |
| Other
income (expense) | |
| (54,282 | ) | |
| (45,365 | ) |
| Total
other income (expense) | |
| (3,793,642 | ) | |
| 121,723 | |
| | |
| | | |
| | |
| Net loss | |
$ | (8,829,996 | ) | |
$ | (6,633,790 | ) |
| Dividends on preferred
stock | |
| (1,355,347 | ) | |
| (2,076,737 | ) |
| Net loss attributable
to common shareholders | |
$ | (10,185,343 | ) | |
$ | (8,710,527 | ) |
| | |
| | | |
| | |
| Net loss per share, basic and diluted | |
$ | (1.97 | ) | |
$ | (8.25 | ) |
| | |
| | | |
| | |
| Weighted average common stock outstanding,
basic and diluted | |
| 5,181,107 | | |
| 1,055,380 | |
The
accompanying notes are an integral part of these condensed financial statements.
AZITRA,
INC.
Statements
of Convertible Preferred Stock and Stockholders’ Deficit
For
the Nine Months Ended September 30, 2023 and 2022
Unaudited
| | |
Series
A Convertible Preferred Stock | | |
Series
A-1 Convertible Preferred Stock | | |
Series
B Convertible Preferred Stock | | |
Common
Stock | | |
Additional
Paid-in- | | |
Accumulated | | |
Total
Stockholders’
Equity | |
| | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Capital | | |
Deficit | | |
(Deficit) | |
Balance
– December 31, 2021 | |
| 205,385 | | |
$ | 3,272,944 | | |
| 380,657 | | |
$ | 14,100,533 | | |
| 391,303 | | |
$ | 16,321,065 | | |
| 1,043,100 | | |
$ | 104 | | |
| 868,163 | | |
$ | (26,634,186 | ) | |
$ | (25,765,919 | ) |
| Stock-based compensation | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 56,983 | | |
| - | | |
| 56,983 | |
| Exercise of stock options | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 888 | | |
| - | | |
| 1,510 | | |
| - | | |
| 1,510 | |
| Net
loss | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (2,326,017 | ) | |
| (2,326,017 | ) |
Balance,
March 31, 2022 | |
| 205,385 | | |
| 3,272,944 | | |
| 380,657 | | |
| 14,100,533 | | |
| 391,303 | | |
| 16,321,065 | | |
| 1,043,988 | | |
| 104 | | |
| 926,656 | | |
| (28,960,203 | ) | |
| (28,033,443 | ) |
| Stock-based compensation | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 53,826 | | |
| - | | |
| 53,826 | |
| Net
loss | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (1,890,153 | ) | |
| (1,890,153 | ) |
Balance,
June 30, 2022 | |
| 205,385 | | |
| 3,272,944 | | |
| 380,657 | | |
| 14,100,533 | | |
| 391,303 | | |
| 16,321,065 | | |
| 1,043,988 | | |
| 104 | | |
| 980,482 | | |
| (30,850,356 | ) | |
| (29,869,770 | ) |
| Stock-based compensation | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 44,329 | | |
| - | | |
| 44,329 | |
| Net
loss | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (2,417,620 | ) | |
| (2,417,620 | ) |
Balance
– September 30, 2022 | |
| 205,385 | | |
$ | 3,272,944 | | |
| 380,657 | | |
$ | 14,100,533 | | |
| 391,303 | | |
$ | 16,321,065 | | |
| 1,043,988 | | |
$ | 104 | | |
$ | 1,024,811 | | |
$ | (33,267,976 | ) | |
$ | (32,243,061 | ) |
Balance
– December 31, 2022 | |
| 205,385 | | |
$ | 3,272,944 | | |
| 380,657 | | |
$ | 14,100,533 | | |
| 391,303 | | |
$ | 16,321,065 | | |
| 1,043,988 | | |
$ | 104 | | |
$ | 1,054,138 | | |
$ | (37,314,552 | ) | |
$ | (36,260,310 | ) |
| Issuance of Series B Convertible Preferred | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | |
| Stock | |
| - | | |
| - | | |
| - | | |
| - | | |
| 23,432 | | |
| 1,124,759 | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 1,124,759 | |
| Stock-based compensation | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 38,794 | | |
| - | | |
| 38,794 | |
| Net loss | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (2,457,179 | ) | |
| (2,457,179 | ) |
Balance,
March 31, 2023 | |
| 205,385 | | |
| 3,272,944 | | |
| 380,657 | | |
| 14,100,533 | | |
| 414,735 | | |
| 17,445,824 | | |
| 1,043,988 | | |
| 104 | | |
| 1,092,932 | | |
| (39,771,731 | ) | |
| (38,678,695 | ) |
| Conversion of convertible notes payable | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 1,846,020 | | |
| 185 | | |
| 9,494,887 | | |
| - | | |
| 9,495,072 | |
| | |
Series
A Convertible Preferred Stock | | |
Series
A-1 Convertible Preferred Stock | | |
Series
B Convertible Preferred Stock | | |
Common
Stock | | |
Additional
Paid-in- | | |
Accumulated | | |
Total
Stockholders’ Equity | |
| | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Capital | | |
Deficit | | |
(Deficit) | |
| Conversion
of preferred stock | |
| (205,385 | ) | |
| (3,272,944 | ) | |
| (380,657 | ) | |
| (14,100,533 | ) | |
| (414,735 | ) | |
| (17,445,824 | ) | |
| 7,707,635 | | |
| 771 | | |
| 34,818,530 | | |
| - | | |
| 34,819,301 | |
| Initial public offering,
net of issuance costs of $1,508,791 | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 1,500,000 | | |
| 150 | | |
| 5,991,209 | | |
| - | | |
| 5,991,359 | |
| Stock-based compensation | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 38,794 | | |
| - | | |
| 38,794 | |
| Net
loss | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (4,429,528 | ) | |
| (4,429,528 | ) |
Balance,
June 30, 2023 | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 12,097,643 | | |
| 1,210 | | |
| 51,436,352 | | |
| (44,201,259 | ) | |
| 7,236,303 | |
| Stock based compensation
expense | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 39,073 | | |
| - | | |
| 39,073 | |
| Net
loss | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (1,943,289 | | |
| (1,943,289 | ) |
Balance
– September 30, 2023 | |
| - | | |
$ | - | | |
| - | | |
$ | - | | |
| - | | |
$ | - | | |
| 12,097,643 | | |
$ | 1,210 | | |
$ | 51,475,425 | | |
$ | (46,144,548 | ) | |
$ | 5,332,087 | |
The
accompanying notes are an integral part of these condensed financial statements.
AZITRA,
INC.
Statements
of Cash Flows
Unaudited
| | |
For
the Nine Months Ended
September 30, | |
| | |
2023 | | |
2022 | |
| Cash flows from operating
activities: | |
| | | |
| | |
| Net loss | |
$ | (8,829,996 | ) | |
$ | (6,633,790 | ) |
| Adjustments to reconcile net loss to net cash
used in operating activities: | |
| | | |
| | |
| Depreciation and amortization | |
| 97,390 | | |
| 93,515 | |
| Amortization of debt discount | |
| - | | |
| 5,895 | |
| Amortization of right-of-use
assets | |
| 219,658 | | |
| 212,470 | |
| Accrued interest on convertible
notes | |
| 165,939 | | |
| 60,877 | |
| Stock based compensation | |
| 116,661 | | |
| 155,138 | |
| Change in fair value of
warrant liability | |
| (9,350 | ) | |
| (420 | ) |
| Change in fair value of
convertible notes | |
| 3,630,100 | | |
| - | |
| Forgiveness of accounts
payable | |
| (56,285 | ) | |
| - | |
| Loss on disposal of property
and equipment | |
| 41,417 | | |
| 7,923 | |
| Impairment of intangible
assets and patent costs | |
| 351,360 | | |
| - | |
| | |
| | | |
| | |
| Changes in operating assets and liabilities: | |
| | | |
| | |
| Accounts receivable | |
| 182,820 | | |
| 75,172 | |
| Prepaid expenses | |
| (249,037 | ) | |
| 45,636 | |
| Other assets | |
| (39 | ) | |
| (39,847 | ) |
| Tax credits receivable | |
| 29,141 | | |
| 48,871 | |
| Accounts payable and accrued
expenses | |
| (143,662 | ) | |
| (1,864 | ) |
| Financing lease liability | |
| 44,206 | | |
| - | |
| Operating lease liability | |
| (213,285 | ) | |
| (202,853 | ) |
| Contract
liabilities | |
| (156,000 | ) | |
| (3,500 | ) |
| Net
cash used in operating activities | |
| (4,778,962 | ) | |
| (6,176,777 | ) |
| | |
| | | |
| | |
| Cash flows from investing
activities: | |
| | | |
| | |
| Purchases of property and
equipment | |
| (22,882 | ) | |
| (23,198 | ) |
| Proceeds from sale of property
and equipment | |
| - | | |
| 4,250 | |
| Capitalization of deferred
patent costs | |
| (208,723 | ) | |
| (117,244 | ) |
| Capitalization of licenses | |
| (13,096 | ) | |
| (57,372 | ) |
| Capitalization
of patent and trademark costs | |
| (13,573 | ) | |
| (46,406 | ) |
| Net
cash used in investing activities | |
| (258,274 | ) | |
| (239,970 | ) |
| | |
| | | |
| | |
| Cash flows from financing
activities | |
| | | |
| | |
| Proceeds from convertible
notes, net of issuance costs | |
| - | | |
| 4,350,000 | |
| Payment of deferred offering
costs | |
| - | | |
| (40,703 | ) |
| Principal payments on finance
leases | |
| (46,452 | ) | |
| - | |
| Proceeds from initial public
offering, net | |
| 5,991,359 | | |
| - | |
| Proceeds
from exercise of stock options | |
| - | | |
| 1,510 | |
| Net
cash provided by financing activities | |
| 5,944,907 | | |
| 4,310,807 | |
| | |
| | | |
| | |
| Net change in cash and cash equivalents | |
| 907,671 | | |
| (2,105,940 | ) |
| | |
| | | |
| | |
| Cash and cash equivalents
at beginning of period | |
| 3,492,656 | | |
| 8,044,262 | |
| Cash and cash equivalents
at end of period | |
$ | 4,400,327 | | |
$ | 5,938,322 | |
| | |
| | | |
| | |
| Supplemental disclosure
of non-cash investing and financing information: | |
| | | |
| | |
| | |
| | | |
| | |
| Obtaining a right-of-use asset in exchange
for lease liability | |
$ | - | | |
$ | 1,418,502 | |
| Obtaining a right-of-use asset in exchange
for financing lease liability | |
$ | 46,452 | | |
$ | - | |
| Conversion of note to common stock | |
$ | 9,495,152 | | |
$ | - | |
| Conversion of note to Series B Convertible
Preferred Stock | |
$ | 1,124,759 | | |
$ | - | |
The
accompanying notes are an integral part of these condensed financial statements.
AZITRA,
INC.
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
For The Nine Months Ended September 30, 2023 and 2022
1.Organization
and Nature of Operations
Azitra,
Inc. was founded on January 2, 2014. It is a synthetic biology company focused on screening and genetically engineering microbes of the
skin. The mission is to discover and develop novel therapeutics to create a new paradigm for treating skin disease. The Company’s
discovery platform is screened for naturally occurring bacterial cells with beneficial effects. These microbes are then genomically sequenced
and engineered to make cellular therapies, recombinant therapeutic proteins, peptides and small molecules for precision treatment of
dermatology diseases. On May 17, 2023, the Company changed its name from “Azitra Inc” to “Azitra, Inc.”
The
Company maintains a location in Montreal, Canada for certain research activities. This location and operations completed there remained
consistent throughout 2022 and into 2023. The Company also opened a manufacturing and laboratory space in Groton, Connecticut during
2021.
Forward
Stock Split, Change in Par Value, and Initial Public Offering
In
June 2023, the Company completed its initial public offering (IPO) in which it issued and sold 1,500,000 shares of its common stock at
a price to the public of $5.00 per share. The shares began trading on the NYSE American on June 16, 2023 under the symbol “AZTR”.
The net proceeds received by the Company from the offering were $6.0 million, after deducting underwriting discounts, commissions and
other offering expenses.
Immediately
prior to the effectiveness of the Company’s registration statement, the Company effected a 7.1-for-1 forward stock split of its
issued and outstanding shares of common stock (the Forward Stock Split). On May 17, 2023, the Company changed the par value of its capital
stock from $0.01 to $0.0001. Accordingly, all share and per share amounts for all periods presented in the accompanying unaudited consolidated
financial statements and notes thereto have been adjusted retroactively, where applicable, to reflect the effect of the Forward Stock
Split. Refer to Note 8 for additional details relating to the Forward Stock Split.
Going
Concern Matters
The
unaudited condensed financial statements have been prepared on the going concern basis, which assumes that the Company will continue
in operation for the foreseeable future and which contemplates the realization of assets and liquidation of liabilities in the normal
course of business. However, management has identified the following conditions and events that created an uncertainty about the ability
of the Company to continue as a going concern. As of and for the nine months ended September 30, 2023, the Company has an accumulated
deficit of $46.1 million, a loss from operations of $5.0 million and used $4.8 million to fund operations. These factors among others
raise substantial doubt about the Company’s ability to continue as a going concern.
Management
plans to continue to raise funds through equity and debt financing to fund operating and working capital needs; however, the Company
will require a significant amount of additional funds to complete the development of its product and to fund additional losses which
the Company expects to incur over the next few years. The Company is still in its clinical phase and therefore does not yet have product
revenue. There can be no assurance that the Company will be successful in securing additional financing, if needed, to meet its operating
needs.
These
conditions and events create an uncertainty about the ability of the Company to continue as a going concern for twelve months from the
date that the financial statements are available to be issued. The financial statements do not include any adjustments that might be
necessary should the Company be unable to continue as a going concern.
Reclassification
Certain
prior period amounts and disclosures have been reclassified to conform to the current period’s financial presentation.
AZITRA,
INC.
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
For The Nine Months Ended September 30, 2023 and 2022
2.
Summary of Significant Accounting Policies
Basis
of Accounting
The
financial statements of the Company are prepared in accordance with United States generally accepted accounting principles (“U.S.
GAAP”).
Unaudited
Interim Financial Information
The
unaudited interim financial statements and related notes have been prepared in accordance with U.S. GAAP for interim financial information,
within the rules and regulations of the United States Securities and Exchange Commission (the “SEC”). Certain information
and disclosures normally included in the annual financial statements prepared in accordance with U.S. GAAP have been condensed or omitted
pursuant to such rules and regulations. The unaudited interim financial statements have been prepared on a basis consistent with the
audited financial statements and in the opinion of management, reflect all adjustments, consisting of only normal recurring adjustments,
necessary for the fair presentation of the results for the interim periods presented and of the financial condition as of the date of
the interim balance sheet. The financial data and the other information disclosed in these notes to the interim financial statements
related to the three and nine months are unaudited. Unaudited interim results are not necessarily indicative of the results for the full
fiscal year. These unaudited interim financial statements should be read in conjunction with the financial statements of the Company
for the year ended December 31, 2022, and notes thereto that are included in the Company’s Registration Statement filed with the
SEC on June 5, 2023.
Use
of Estimates
The
preparation of the financial statement in conformity with accounting principles generally accepted in the United States of America requires
management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the balance sheet.
While management believes the estimates and assumptions used in the preparation of the financial statement are appropriate, actual results
could differ from those estimates.
Cash
and Cash Equivalents
For
purposes of the balance sheets and statements of cash flows, the Company considers all cash on hand, demand deposits and all highly liquid
investments with original maturities of three months or less to be cash equivalents.
Property
and Equipment
Property
and equipment are recorded at cost. Depreciation is computed using the straight-line method over the estimated useful lives, which range
from 3 to 10 years. Expenditures for maintenance and repairs, which do not extend the economic useful life of the related assets, are
charged to operations as incurred. Gains or losses on disposal of property and equipment are reflected in the statements of operations
in the period of disposal.
Accounts
Receivable
The
Company carries its accounts receivable at cost less an allowance for doubtful accounts. On a periodic basis, the Company evaluates its
accounts receivable and establishes an allowance for doubtful accounts based on a history of past write-offs, collections and current
conditions. There was no allowance for doubtful accounts at September 30, 2023 and December 31, 2022. Accounts receivable are written
off when deemed uncollectible. Recoveries of accounts receivable previously written off are recorded when received.
AZITRA,
INC.
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
For The Nine Months Ended September 30, 2023 and 2022
Deferred
Offering Costs
The
Company capitalized deferred offering costs, which primarily consisted of direct, incremental legal, professional, accounting, and other
third-party fees relating to the Company’s initial public offering. In June 2023, the Company consummated its IPO and recorded
such amounts against the gross proceeds of its IPO within the statements of stockholders’ equity during the nine months ended September
30, 2023.
Leases
In
February 2016, the Financial Accounting Standards Board (“FASB”) issued ASU No. 2016-02, Leases (“Topic 842”).
ASU 2016-02 requires lessees to present right-of-use (“ROU”) assets and lease liabilities on the balance sheet for all leases
with terms longer than 12 months. See Note 2 - Recently Adopted Accounting Pronouncements.
In
calculating the effect of ASU 2016-02, the Company elected the transition method thereby not restating comparable periods. The Company
elected to account for non-lease components as part of the lease component to which they relate. Lease accounting involves significant
judgments, including making estimates related to the lease term, lease payments, and discount rate. In accordance with the guidance,
the Company recognized ROU assets and lease liabilities for all leases with a term greater than 12 months. Leases are classified as either
operating or financing leases based on the economic substance of the agreement.
The
Company has operating leases for buildings. Currently, the Company has 3 operating leases with a ROU asset and lease liability totaling
$1,418,502 as of January 1, 2022. The basis, terms and conditions of the leases are determined by the individual agreements. The Company’s
option to extend certain leases ranges from 36 - 52 months. All options to extend have been included in the calculation of the ROU asset
and lease liability. The leases do not contain residual value guarantees, restrictions, or covenants that could incur additional financial
obligations to the Company. There are no subleases, sale-leaseback, or related party transactions.
At
September 30, 2023, the Company had operating right-of-use assets with a net value of $899,619 and current and long-term operating lease
liabilities of $301,423 and $613,572, respectively.
In
2023, the Company entered into a lease for the use of certain equipment that is classified as a finance lease. The finance lease has
a term of 36 months. At September 30, 2023, the Company had financing right-of-use assets with a net value of $43,872 and current and
long-term operating lease liabilities of $14,254 and $29,952, respectively.
Intangible
Assets
Intangible
assets consist of trademarks and patents. All costs directly related to the filing and prosecution of patent and trademark applications
are capitalized. Patents are amortized over their respective remaining useful lives upon formal approval. Trademarks have an indefinite
life.
The
Company accounts for other indefinite life intangible assets in accordance ASC Topic 350, Goodwill and Other Intangible Assets
(ASC 350). ASC 350 requires that intangible assets that have indefinite lives are required to be tested at least annually for impairment
or whenever events or changes in circumstances indicate that the carrying amount may not be recoverable. Intangible assets that have
finite lives will continue to be amortized over their useful lives. Impairment losses relating to license agreements were $78,606 during
the nine months ended September 30, 2023 and $0 during the nine months ended September 30, 2022.
Deferred
Patent Costs
Deferred
patent costs represent legal and filing expenses incurred related to the submission of patent applications for patents pending approval.
These deferred costs will begin to be amortized over their estimated useful lives upon the formal approval of the patent. If the patent
is not issued, the costs associated with the patent will be expensed in the year the patent was rejected. Impairment losses relating
to deferred patent costs were $272,754 during the nine months ended September 30, 2023 and $0 in the nine months ended September 30,
2022.
AZITRA,
INC.
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
For The Nine Months Ended September 30, 2023 and 2022
Impairment
of Long-Lived Assets
In
accordance with ASC Topic 360-10, Accounting for the Impairment or Disposal of Long-Lived Assets (ASC 360-10), the Company’s policy
is to review its long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an
asset may not be recoverable. In connection with this review, the Company also reevaluates the periods of depreciation for these assets.
The Company recognizes an impairment loss when the sum of the undiscounted expected future cash flows from the use and eventual disposition
of the asset is less than its carrying amount. If an asset is considered to be impaired, the impairment to be recognized is measured
by the amount by which the carrying amount of the asset exceeds the fair value of the asset, which is determined using the present value
of the net future operating cash flows generated by the asset.
Convertible
Debt and Warrant Accounting
Warrants
The
Company accounts for warrants as either equity-classified or liability-classified instruments based on an assessment of the warrant’s
specific terms and applicable authoritative guidance in ASC 480, Distinguishing Liabilities from Equity (“ASC 480”)
and ASC 815, Derivatives and Hedging (“ASC 815”). The assessment considers whether the warrants are freestanding financial
instruments pursuant to ASC 480, meet the definition of a liability pursuant to ASC 480, and whether the warrants meet all of the requirements
for equity classification under ASC 815, including whether the warrants are indexed to the Company’s own common stock, among other
conditions for equity classification. This assessment, which requires the use of professional judgment, is conducted at the time of warrant
issuance and as of each subsequent quarterly period end date while the warrants are outstanding.
For
issued warrants that meet all of the criteria for equity classification, the warrants are required to be recorded as a component of additional
paid-in capital at the time of issuance. For issued warrants that do not meet all the criteria for equity classification, the warrants
are required to be recorded at their initial fair value on the date of issuance, and each balance sheet date thereafter. Changes in the
estimated fair value of the warrants are recognized as a non-cash gain or loss on the statements of operations under Other Income/loss.
Convertible
debt
When
the Company issues debt with a conversion feature, it first assesses whether the debt should be accounted for in accordance with ASC
480 - Distinguishing Liabilities from Equity. If the debt does not meet the criteria of an ASC 480 liability, the note’s conversion
features require bifurcation in accordance with ASC 815 - Derivatives and Hedging. If the Company determines the embedded conversion
feature requires bifurcation in accordance with ASC 815, the Company also considers if it can elect the fair value option. If the fair
value option is elected, the Company records the note at its initial fair value with any subsequent changes in fair value recorded in
earnings. As noted in Note 7, the Company has elected the fair value option for the 2022 Convertible Notes and will record the notes
at their initial fair values with any subsequent changes in fair value recorded in earnings. The Convertible Notes were converted into
the Company’s common stock on the Closing Date of the Company’s IPO.
Convertible
Preferred Stock
As
the Convertible Preferred stockholders have liquidation rights in the event of a deemed liquidation event that, in certain situations,
are not solely within the control of the Company and would require the redemption of the then-outstanding Convertible Preferred Stock,
the Company classifies the Convertible Preferred Stock in mezzanine equity on the balance sheet.
As
noted in Note 8, at the Closing Date of the Company’s IPO, the Convertible Preferred stock converted into shares of the Company’s
common stock.
AZITRA,
INC.
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
For The Nine Months Ended September 30, 2023 and 2022
Revenue
The
Company follows the five steps to recognize revenue from contracts with customers under ASC 606, Revenue from Contracts with Customers
(“ASC 606”), which are:
| ● | Step
1: Identify the contract(s) with a customer |
| ● | Step
2: Identify the performance obligations in the contract |
| ● | Step
3: Determine the transaction price |
| ● | Step
4: Allocate the transaction price to the performance obligations in the contract |
| ● | Step
5: Recognize revenue when (or as) a performance obligation is satisfied |
The
Company generates service revenue through a joint development agreement with a research partner. The Company recognizes revenue related
to the research and development aspects of the agreement over time using the input method as work is performed on the contract.
The
Company also generates grant revenue, which represents monies received on contracts with various federal agencies and nonprofit research
institutions for general research conducted by the Company to further their product development and are therefore considered contributions
to the Company. The contracts are generally for periods of one year or more and can be cancelled by either party. The Company concluded
that the grant arrangements do not meet the criteria to be treated as a collaborative arrangement under FASB ASC Topic 808 as the Company
is the only active participant in the arrangement. The grant arrangements also do not meet the criteria for revenue recognition under
Topic 606, as the U.S. Government would not meet the definition of a customer.
Amounts
earned under these grant contracts are recorded as a reduction to research and development expense when eligible expenses are incurred
and the right to payment is realizable or realized and earned. The Company believes this policy is consistent with Topic 606, to ensure
that recognition reflects the transfer of promised goods or services to customers in an amount that reflects the consideration that the
Company expects to be entitled to in exchange for those goods or services, even though there is no exchange as defined in Topic 606.
Additionally, the Company has determined that the recognition of amounts received as costs are incurred and amounts become realizable
is analogous to the concept of transfer of control of a service over time under Topic 606.
Receipts
of grant awards in advance, which are payable back to the funding agency if not used in accordance with conditions in the grants related
to allowable costs or receipt of funding from research partners related to service revenue arrangements before work is performed on the
contract, are classified as contract liabilities in the accompanying balance sheets.
Research
and Development
The
Company accounts for research and development costs in accordance with Accounting Standards Codification (ASC) subtopic 730-10, Research
and Development. Accordingly, internal research and development costs are expensed as incurred. Research and development costs consist
of costs related to labor, materials and supplies. Research and development costs incurred were $2,188,795 during the nine months ended
September 30, 2023. Research and development costs incurred were $4,425,195 during the nine months ended September 30, 2022.
At
September 30, 2023 and December 31, 2022, the Company has a state tax credit receivable of $32,459 for pending refunds related to the
selling of research and development tax credits back to the State of Connecticut. At September 30, 2023 and December 31, 2022, the Company
has $0 and $28,925, respectively for pending refunds related to Canadian Scientific Research and Experimental Development (SRED) credits.
At September 30, 2023 and December 31, 2022, the Company has also recorded $8,066 and $8,282, respectively, related to refunds of Canadian
Goods and Services Tax (GST) and Quebec Sales Tax (QST). Receipts of refunds are recorded in other income on the statements of operations.
AZITRA,
INC.
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
For The Nine Months Ended September 30, 2023 and 2022
Stock-Based
Compensation
The
Company accounts for stock-based compensation in accordance with ASC 718, Compensation-Stock Compensation (ASC 718). ASC 718 requires
employee stock options and rights to purchase shares under stock participation plans to be accounted for at fair value. ASC 718 requires
that compensation costs related to share-based payment transactions be recognized as operating expenses in the financial statements.
Under this method, compensation costs for all awards granted or modified are measured at estimated fair value at date of grant and are
included as compensation expense over the vesting period during which an employee provides service in exchange for the award. For awards
with a performance condition that affects vesting, the Company recognizes compensation expense when it is determined probable that the
performance condition will be achieved. The Company recognized the effect of forfeitures when the forfeitures occur.
The
Company uses a Black-Scholes option pricing model to determine fair value of its stock options. The Black-Scholes model includes various
assumptions, including the value of the underlying common stock, the expected life of stock options, the expected volatility and the
expected risk-free interest rate. These assumptions reflect the Company’s best estimates, but they involve inherent uncertainties
based on market conditions generally outside of the control of the Company. As a result, if other assumptions had been used, stock-based
compensation cost could have been materially impacted. Furthermore, if the Company uses different assumptions for future grants, stock-based
compensation cost could be materially impacted in future periods.
The
Company accounts for equity instruments issued to non-employees in accordance with the provisions of ASC 718 as updated by Accounting
Standards Update (ASU) No. 2018-07, Improvements to Nonemployee Share-Based Payment Accounting, which expands the scope of ASC
718 to include share-based payment transactions to non-employees.
The
following assumptions are used in valuing options issued using the Black-Scholes option pricing model:
Expected
Volatility. The expected volatility of the Company’s shares is estimated based on the average volatility of peer companies.
Expected
Term. The expected term of options is estimated using the simplified method which is based on the vesting period and contractual
term for each grant, or for each vesting-tranche for awards with graded vesting.
Underlying
Common Stock Value. The underlying common stock value of the Company’s shares is estimated by a third-party valuation expert
up until the Company’s IPO, at which time the Company utilized its trading price on the NYSE American on the date of grant.
Risk-free
Interest Rate. The Company bases the risk-free interest rate on the implied yield available on a U.S. Treasury note with terms equal
to the expected term of the underlying grant.
Dividend
Yield. The Black-Scholes valuation model calls for a single expected dividend yield as an input. The Company has not paid dividends
on Common stock in the past nor does it expect to pay dividends on Common stock in the near future. As such, the Company uses a dividend
yield percentage of zero.
Income
Taxes
The
Company uses the liability method of accounting for income taxes, as set forth in ASC 740, Accounting for Income Taxes. Under this method,
deferred tax assets and liabilities are recognized for the expected future tax consequence of temporary differences between the carrying
amounts and the tax basis of assets and liabilities and net operating loss carry forwards, all calculated using presently enacted tax
rates.
Management
has evaluated the effect of ASC guidance related to uncertain income tax positions and concluded that the Company has no significant
financial statement exposure to uncertain income tax positions at September 30, 2023 and December 31, 2022. The Company’s income
tax returns have not been examined by tax authorities through December 31, 2022.
AZITRA,
INC.
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
For The Nine Months Ended September 30, 2023 and 2022
Fair
Value Measurements
The
Company carries certain liabilities at fair value on a recurring basis. A fair value hierarchy that consists of three levels is used
to prioritize the inputs to fair value valuation techniques:
| ● | Level
1 - Inputs are based upon observable or quoted prices for identical instruments traded in
active markets. |
| ● | Level
2 - Inputs are based upon quoted prices for similar instruments in active markets, quoted
prices for identical or similar instruments in markets that are not active, and model-based
valuation techniques for which all significant assumptions are observable in the market or
can be corroborated by observable market data for substantially the full term of the assets
or liabilities. |
| ● | Level
3 - Inputs are generally unobservable and typically reflect management’s estimates
of assumptions that market participants would use in pricing the asset or liability. The
fair values are therefore determined using model-based techniques that include option pricing
models, discounted cash flow models, and similar techniques. |
In
determining fair value, the Company utilizes valuation techniques that maximize the use of observable inputs and minimize the use of
unobservable inputs to the extent possible as well as considers counterparty credit risk in its assessment of fair value.
Financial
Instruments
The
Company’s financial instruments are primarily comprised of accounts receivable, accounts payable, accrued liabilities, and long-term
debt. For accounts receivable, accounts payable and accrued liabilities, the carrying amount approximates fair value due to the short-term
maturities of such instruments. The estimated fair value of the Company’s long-term debt approximates carrying value.
Recent
Accounting Pronouncements
In
February 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) 2016-02,
Leases (Topic 842). This ASU requires a lessee to recognize a right-of-use asset and a lease liability under most operating leases in
its balance sheet. The ASU is effective for annual and interim periods beginning after December 15, 2021. The Company adopted ASU 2016-02
on January 1, 2022. See Note 13 - Operating Leases.
In
December 2019, the FASB issued ASU No. 2019-12, Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes.
This standard simplifies the accounting for income taxes through the removal of various exceptions previously provided, as well as providing
additional reporting requirements for income taxes. The ASU is effective for the Company on January 1, 2022. The Company has adopted
this standard effective January 1, 2022, which did not have a material impact to the financial statements.
In
August 2020, the FASB issued ASU No. 2020-06, Debt - Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and
Hedging - Contracts in Entity’s Own Equity (Subtopic 815-40), which simplifies the accounting for certain financial instruments
with characteristics of liabilities and equity, including convertible instruments and contracts on an entity’s own equity. This
standard will be effective for the Company on January 1, 2024, with early adoption permitted (but no earlier than fiscal years beginning
after December 15, 2020). The Company has adopted this standard effective January 1, 2021, which did not have a material impact to the
financial statements.
AZITRA,
INC.
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
For The Nine Months Ended September 30, 2023 and 2022
Management
does not believe that any other recently issued, but not yet effective, accounting standards could have a material effect on the accompanying
financial statements. As new accounting pronouncements are issued, the Company will adopt those that are applicable under the circumstances.
3.
Employee Retention Credit
The
CARES Act provides an employee retention credit (“CARES Employee Retention credit”), which is a refundable tax credit against
certain employment taxes of up to $5,000 per employee for eligible employers. The tax credit is equal to 50% of qualified wages paid
to employees during a quarter, capped at $10,000 of qualified wages per employee through December 31, 2020. Additional relief provisions
were passed by the United States government, which extend and slightly expand the qualified wage caps on these credits through September
30, 2021. Based on these additional provisions, the tax credit is now equal to 70% of qualified wages paid to employees during a quarter,
and the limit on qualified wages per employee has been increased to $10,000 of qualified wages per quarter. In April 2022, the Company
determined it qualified for the tax credit under the CARES Act and recorded a receivable for $229,813 and recognized the amounts as other
income on the statement of operations. The Company received full payment for the amount in September 2022.
4.
Property and Equipment
Property
and equipment consisted of the following at September 30, 2023 and December 31, 2022:
| | |
September
30, 2023 | | |
December
31, 2022 | |
| Laboratory equipment | |
$ | 1,008,063 | | |
$ | 1,034,579 | |
| Computers and office equipment | |
| 30,825 | | |
| 30,825 | |
| Furniture and fixtures | |
| 24,316 | | |
| 24,316 | |
| Leasehold improvements | |
| 28,855 | | |
| 28,855 | |
| Building equipment | |
| 14,932 | | |
| 14,932 | |
| Total property and equipment | |
| 1,106,991 | | |
| 1,133,507 | |
| Less accumulated depreciation
& amortization | |
| (370,568 | ) | |
| (286,549 | ) |
| Total property, plant,
and equipment, net | |
$ | 736,423 | | |
$ | 846,958 | |
Depreciation
expense was $92,000 for the nine months ended September 30, 2023. Depreciation expense was $90,165 for the nine months ended September
30, 2022.
For
the nine months ended September 30, 2023 there was $41,417 of loss on disposal of property and equipment recorded. For the nine months
ended September 30, 2022 there was $7,923 of loss on disposal of property and equipment recorded.
5.
Intangible Assets
Intangible
assets consisted of the following at September 30:
2023:
| | |
Estimated
Useful
Life | |
Gross
Amount | | |
Accumulated
Amortization | | |
Impairment | | |
Net
Amount | |
| Trademarks | |
Indefinite | |
$ | 56,409 | | |
$ | - | | |
$ | - | | |
$ | 56,409 | |
| Patents | |
17 years | |
| 119,361 | | |
| 13,530 | | |
| - | | |
| 105,831 | |
| License agreement | |
17 years | |
| 78,606 | | |
| - | | |
| 78,606 | | |
| - | |
| Intangible assets | |
| |
$ | 254,376 | | |
$ | 13,530 | | |
$ | 78,606 | | |
$ | 162,240 | |
2022:
AZITRA,
INC.
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
For The Nine Months Ended September 30, 2023 and 2022
| | |
Estimated
Useful
Life | |
Gross
Amount | | |
Accumulated
Amortization | | |
Impairment | | |
Net
Amount | |
| Trademarks | |
Indefinite | |
$ | 53,999 | | |
$ | - | | |
$ | - | | |
$ | 53,999 | |
| Patents | |
17 years | |
| 108,198 | | |
| 8,140 | | |
| - | | |
| 100,058 | |
| License agreement | |
17 years | |
| 65,510 | | |
| - | | |
| - | | |
| 65,510 | |
| Intangible assets | |
| |
$ | 227,707 | | |
$ | 8,140 | | |
$ | - | | |
$ | 219,567 | |
During
the nine months ended September 30, 2023, amortization expense related to intangible assets was $5,390. During the nine months ended
September 30, 2022, amortization expense related to intangible assets was $3,350.
6.
Accrued Expenses
Accrued
expenses consisted of the following at September 30, 2023 and 2022:
| | |
September
30, 2023 | | |
December
31, 2022 | |
| Accrued expenses: | |
| | | |
| | |
| Employee payroll and bonuses | |
$ | 272,950 | | |
$ | 371,010 | |
| Vacation | |
| 57,963 | | |
| 27,082 | |
| Research and development projects | |
| 130,233 | | |
| 316,389 | |
| Interest | |
| - | | |
| 223,792 | |
| Professional fees | |
| 255,794 | | |
| 24,502 | |
| Other | |
| 3,155 | | |
| 31,186 | |
| Total accrued expenses | |
$ | 720,095 | | |
$ | 993,961 | |
The
Company accrues expenses related to development activities performed by third parties based on an evaluation of services received and
efforts expended pursuant to the terms of the contractual arrangements. Payments under some of these contracts depend on research and
non-clinical trial milestones. There may be instances in which payments made to the Company’s vendors will exceed the level of
services provided and result in a prepayment of expense. In accruing service fees, the Company estimates the period over which services
will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level
of effort varies from the estimate, the Company will adjust the accrual or prepaid expense accordingly. The Company has not experienced
any material differences between accrued costs and actual costs incurred since its inception.
7.
Convertible Debt
In
September 2022, the Company entered into a Convertible Note Purchase Agreement (the Agreement) to issue up to $4,500,000 convertible
promissory notes. On the same day, the Company entered into convertible promissory notes (2022 Convertible Notes) with three investors
totaling $4,350,000. The 2022 Convertible Notes mature on January 13, 2023 or the occurrence of an Event of Default (as defined) and
bear interest at a rate of 8% per annum which shall accrue but is not due and payable until conversion or full repayment of outstanding
principal. The principal and interest outstanding under the 2022 Convertible Notes is automatically converted a) upon the closing of
a Qualified Financing resulting in gross proceeds to the Company of at least $20 million into securities issued in connection with the
Qualified Financing, at a discount of 30% per share; b) upon the closing of a Change of Control event into shares of capital stock of
the Company or Series B preferred stock; and c) upon the closing of a Public Company Event, into shares of capital stock being issued
to investors equal to two-times (2x) the amount of the outstanding principal and accrued interest then outstanding divided by the public
offering price per share. The principal and interest outstanding under the 2022 Convertible Notes is convertible, at the option of the
holders, at the maturity date into a new class of Company’s Preferred Stock (Series C Preferred) equal to the quotient of the outstanding
principal amount plus interest divided by the Capped Price, which is defined as the price per share equal to the Valuation Cap of $30
million divided by the Company Capitalization, as defined in the Agreement.
AZITRA,
INC.
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
For The Nine Months Ended September 30, 2023 and 2022
In
February 2023, the 2022 Convertible Notes were amended to extend the maturity date to March 31, 2023 and to change the conversion price
upon a Qualified Financing or Change in Control event to $30 million divided by the number of shares of the Company’s common stock
issued and outstanding, on a fully diluted basis, immediately prior to the close of the Qualified Financing or Change in Control event.
During
April and June 2023, the 2022 Convertible Notes were further amended to extend the maturity date to September 30, 2023 and allow for
the sale of additional notes of $500,000 for a total aggregate principal of $4,850,000.
Effective
June 21, 2023, the 2022 Convertible Notes were converted to 1,846,020 shares of the Company’s common stock equal to $9,494,887.
Upon conversion, the Company recorded a change in fair value of $3,630,100 for the nine months ended September 30, 2023, which was recognized
as a non-cash change in fair value in Other Income (Expense) on the statement of operations.
The
Company accounts for the 2022 Convertible Notes under ASC 815. Under 815-15-25, the election can be at the inception of a financial instrument
to account for the instrument under the fair value option under ASC 825. The Company has made such election for the 2022 Convertible
Notes. Using the fair value option, the convertible promissory note is to be recorded at its initial fair value on the date of issuance,
and each balance sheet date thereafter. The Company evaluates the change based on the conversion price at the current market value. When
recognized, changes in the estimated fair value of the notes are recognized as a non-cash gain or loss in Other Income (Expense) on the
statements of operations.
Effective
January 5, 2021, the Company entered into a Note Purchase Agreement to issue up to $2,000,000 of convertible promissory notes. On the
same date, the Company entered into a convertible promissory note (2021 Convertible Note) with one investor for $1,000,000. The 2021
Convertible Note bears interest at a rate of 6% per annum and is due and payable in full on January 5, 2023. The 2021 Convertible Note
automatically converts upon a qualified equity financing, as defined in the note agreement to the number of shares equal to all principal
and accrued interest divided by the conversion price of $48.00, which is subject to adjustment as defined in the note agreement. The
2021 Convertible Note is also optionally convertible as defined in the note agreement for certain non-qualified financing, a change in
control, or upon the maturity date of the 2021 Convertible Note. The Company incurred issuance costs of $15,613 related to the 2021 Convertible
Note, which has been recorded as a debt discount and will be amortized over the term of the 2021 Convertible Note.
In
January 2023, the Company elected to convert the 2021 Convertible Note, including interest accrued but not yet paid of $124,759 at a
conversion price of $48.00 into 23,432 shares of its Series B Preferred Stock in accordance with the terms outlined in the Note Purchase
Agreement.
The
Company evaluated the terms and conditions of the Note Purchase Agreement related to the 2021 Convertible Note in order to assess the
accounting considerations under ASC 480 - Distinguishing Liabilities from Equity, and ASC 815 - Derivatives and Hedging. The Company
determined the Convertible Note does not meet any of the criteria to be accounted pursuant to an ASC 480 liability. The Company also
assessed the embedded features pursuant to the guidance in ASC 815 and determined the embedded features do not meet any of the criteria
for bifurcation.
Convertible
notes payable consisted of the following at:
| | |
September
30, 2023 | | |
December
31, 2022 | |
| 2021 Convertible
Note | |
$ | - | | |
$ | 1,000,000 | |
| 2022
Convertible Notes | |
| - | | |
| 5,600,000 | |
| Total convertible notes | |
$ | - | | |
$ | 6,600,000 | |
There
was $0 amortized related to the debt issuance costs during the nine months ended September 30, 2023. There was $5,904 amortized related
to the debt issuance costs during the nine months ended September 30, 2022. Interest accrued on the convertible notes was $0 and $223,792
at September 30, 2023 and December 31, 2022, respectively.
AZITRA,
INC.
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
For The Nine Months Ended September 30, 2023 and 2022
8.
Stockholders’ Equity
On
May 17, 2023, the Company effected a 7.1-for-1 forward stock split (the “Forward Stock Split”) of its issued and outstanding
shares of common stock and a proportional adjustment to the existing conversion ratios for each series of the Company’s preferred
stock. The par value of the common stock was adjusted as a result of the Forward Stock Split from $0.01 to $0.0001 and the authorized
shares were increased to 100,000,0000 shares of common stock in connection with the Forward Stock Split. Fractional shares resulting
from the Forward Stock Split were rounded down to the next whole share and in lieu of any fractional shares the Company will pay a cash
amount to the holder of such fractional share. The accompanying financial statements and notes to the financial statements give retroactive
effect to the Forward Stock Split for all periods presented. Shares of common stock underlying outstanding stock-based awards and other
equity instruments were proportionately increased and the respective per share value and exercise prices, if applicable, were proportionately
decreased in accordance with the terms of the agreements governing such securities.
Common
Stock
At
September 30, 2023 and December 31, 2022, per the Company’s amended and restated Certificate of Incorporation, the Company was
authorized to issue 100,000,000 shares of $0.0001 par value common stock.
The
Company had 12,097,643 and 1,043,988 shares of common stock issued and outstanding as of September 30, 2023 and December 31, 2022, respectively.
Each
share of common stock entitles the holder to one vote on all matters submitted to a vote of the Company’s stockholders and the
holders of the Common Stock are entitled to elect one director of the Corporation.
The
Company currently has 1,611,991 shares of common stock reserved for future issuance for the potential exercise of stock options and warrants
outstanding at September 30, 2023.
Preferred
Stock
At
September 30, 2023 and December 31, 2022, per the Company’s amended and restated Certificate of Incorporation, the Company has
authorized 1,437,150 shares of $0.0001 par value preferred stock.
In
January 2023, the Company issued 23,432 shares of its Series B Preferred Stock related to conversion of the 2021 Convertible Note at
a conversion price of $48.00 per share (see Note 7).
The
Series A, Series A-1, and Series B Preferred Stock have the following rights, preferences and privileges:
Conversion
The
preferred stock is convertible, at the option of the holder, into common shares based upon a predefined formula. A holder of preferred
stock may convert such shares into common shares at any time. For purpose of conversion, the initial conversion price is $16.25 per share
(original issue price) for Series A Preferred Stock, $37.50 per share (original issue price) for Series A-1 Preferred Stock, and $43.45
per share (original issue price) for Series B Preferred Stock, and is subject to adjustment as described in the Certificate of Incorporation.
Preferred stock will automatically convert into common shares upon the earlier of (a) an initial public offering with gross proceeds
in excess of $100,000,000 or (b) the date and time, or the occurrence of an event, specified by vote or written consent of the required
preferred stock shareholders, all outstanding Series A, Series A-1, and Series B Preferred Stock shall automatically convert into common
shares, at the then effective conversion rate.
AZITRA,
INC.
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
For The Nine Months Ended September 30, 2023 and 2022
Upon
the Company’s IPO in June 2023, all of the outstanding preferred stock converted to common stock, resulting in the issuance of
1,458,233, 2,964,849, and 3,284,553 shares of common stock in exchange for outstanding Series A, Series A-1, and Series B Preferred Stock,
respectively. There was no gain or loss upon conversion.
Voting
Rights
The
holders of the Series A, Series A-1, and Series B Preferred Stock are entitled to vote on any matter presented to the stockholders of
the Corporation for their action or consideration at any meeting of stockholders of the Corporation (or by written consent of stockholders
in lieu of meeting), each holder of outstanding shares of preferred stock shall be entitled to cast the number of votes equal to the
number of whole shares of Common Stock into which the shares of preferred stock held by such holder are convertible as of the record
date for determining stockholders entitled to vote on such matter. The holders of the Series A and Series A-1 Preferred Stock are each
entitled to elect one director of the Corporation. The holders of the Series B Stock are entitled to elect two members of the Board.
Each class of preferred stock can remove from office such directors and to fill any vacancy caused by the resignation, death or removal
of such directors under certain circumstances as described in the Certificate of Incorporation.
Dividends
The
holders of Series A Preferred Stock are entitled to receive dividends at a rate of 8% per annum of the Series A original issue price
of $16.25 per share on each outstanding share of Series A Preferred Stock (subject to appropriate adjustment in the event of any stock
dividend, stock split, combination or other similar recapitalization with respect to the Series A Preferred Stock). Dividends accumulate
from the original date of issuance of the Series A Preferred Stock, are cumulative and are payable upon declaration of the Board of Directors
or liquidation of the Company. At September 30, 2023, there were no cumulative dividends on Series A Preferred Stock as there was no
longer any Series A Preferred Stock outstanding.
The
holders of Series A-1 Stock are entitled to receive dividends at a rate of 8% per annum of the Series A-1 original issue price of $37.50
per share on each outstanding share of Series A-1 (subject to appropriate adjustment in the event of any stock dividend, stock split,
combination or other similar recapitalization with respect to the Series A-1 Preferred Stock). Dividends are cumulative and are payable
upon declaration of the Board of Directors or liquidation of the Company. At September 30, 2023, there were no cumulative dividends on
Series A-1 Preferred Stock as there was no longer any Series A-1 Preferred Stock outstanding.
The
holders of Series B Stock are entitled to receive dividends at a rate of 8% per annum of the Series B original issue price of $43.45
per share on each outstanding share of Series B (subject to appropriate adjustment in the event of any stock dividend, stock split, combination
or other similar recapitalization with respect to the Series B Preferred Stock). Dividends are cumulative and are payable upon declaration
of the Board of Directors or liquidation of the Company. At September 30, 2023, there were no cumulative dividends on Series B Preferred
Stock as there was no longer any Series B Preferred Stock outstanding.
Liquidation
In
the event of any liquidation, dissolution or winding up of the Company, the holders of the preferred stock are entitled to receive, prior
to and in preference to the holders of the common shares, an amount equal to the Series A, Series A-1, or Series B Preferred Stock original
issue price, plus declared and/or accrued but unpaid dividends. In the event of any such liquidation event, after the payment of all
preferential amounts required to be paid to the holders of shares of preferred stock, the remaining assets of the Corporation available
for distribution to its stockholders shall be distributed among the holders of the shares of preferred stock and Common Stock, pro rata
based on the number of shares held by each such holder, treating for this purpose all such securities as if they had been converted into
Common Stock pursuant to the terms of the Certificate of Incorporation immediately prior to such liquidation event.
AZITRA,
INC.
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
For The Nine Months Ended September 30, 2023 and 2022
9.
Warrants
The
Company issued warrants to purchase 6,745 shares of common stock in 2018 in conjunction with convertible debt financing that have a redemption
provision providing the holder the right to have the Company redeem all or any portion of the warrant (or shares it has converted into)
at a purchase price equal to the fair market value of the shares as determined by the board of directors or an independent appraiser.
As a result of this redemption provision, the warrants have been classified as a liability in the financial statements based on ASC 480
- Distinguishing Liabilities from Equity. These warrants have an exercise price of $0.48 per share and a term of 10 years. The warrants
are marked to market each reporting period. The fair value is $60,933 and $70,283 at September 30, 2023 and December 31, 2022, respectively.
The
Company also issued warrants in 2016 and 2019 which did not meet the criteria under ASC 480 to be classified as a liability, and instead
meet equity classification criteria. The 11,466 warrants issued in 2016 expired upon the initial public offering in June 2023.
The
following table summarizes information about warrants outstanding at September 30, 2023:
| |
|
|
|
|
|
|
Warrants
Outstanding |
|
|
|
Warrant
Exercisable |
|
| Year
Granted |
|
|
Exercise
Price |
|
|
|
Number
of Warrants at 9/30/2023 |
|
|
|
Weight
Average Remaining Contractual Life |
|
|
|
Weight
Average Exercise Price |
|
|
|
Number
of Warrants at 9/30/2023 |
|
|
|
Weighted
Average Remaining Contractual Life |
|
|
|
Weighted
Average Exercise Price |
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 2018 |
|
$ |
0.48 |
|
|
|
47,890 |
|
|
|
4.5
years |
|
|
$ |
0.48 |
|
|
|
47,890 |
|
|
|
4.5
years |
|
|
$ |
0.48 |
|
| 2019 |
|
$ |
5.28 |
|
|
|
215,846 |
|
|
|
2.4
years |
|
|
$ |
5.28 |
|
|
|
215,846 |
|
|
|
2.4
years |
|
|
$ |
5.28 |
|
| 2023 |
|
$ |
6.25 |
|
|
|
60,000 |
|
|
|
4.7
years |
|
|
$ |
6.25 |
|
|
|
60,000 |
|
|
|
4.7
years |
|
|
$ |
6.25 |
|
| |
|
|
|
|
|
|
323,736 |
|
|
|
|
|
|
$ |
4.75 |
|
|
|
323,736 |
|
|
|
|
|
|
$ |
4.75 |
|
10.
Stock Options
In
March 2023, the Company’s Board of Directors and stockholders approved the 2023 Stock Incentive Plan (“2023 Plan”).
The 2023 Plan allows the Committee to grant up to 2,000,000 shares of Common Stock in the form of incentive and non-statutory stock options,
restricted stock awards, restricted stock units, and other stock-based awards to employees, directors, and non-employees. As of September
30, 2023, options to purchase 40,000 shares of common stock had been granted and were outstanding under the 2023 Plan and 1,960,000 shares
of common stock were available for grant under the plan.
During
2016, the Company established the Azitra Inc. 2016 Stock Incentive Plan (the Plan) which provides for the granting of stock options and
restricted shares to the Company’s employees, officers, directors, advisors and consultants. As of September 30, 2023, options
to purchase 1,248,255 shares of common stock had been granted and were outstanding under the 2016 Plan and 242,345 shares of common stock
were available for grant under the plan.
During
the nine months ended September 30, 2023 the Company granted stock options to purchase 40,000 shares of common stock and during the nine
months ended September 30, 2022 the Company did not grant any stock options under this plan. During the nine months ended September 30,
2023, the Company recognized stock compensation expense of $116,661, relating to the issuance of service-based stock options. During
the nine months ended September 30, 2022, the Company recognized stock compensation expense of $155,138, relating to the issuance of
service-based stock options. At September 30, 2023, there was $361,288 of unamortized compensation expense that will be amortized over
the remaining vesting period. At September 30, 2023 and 2022, there were 13,120 performance-based options outstanding with a fair value
of $109,551. During the nine months ended September 30, 2023 and 2022, the Company did not recognize any compensation expense for performance-based
options. The Company determined the options qualified as plain vanilla under the provisions of SAB 107 and the simplified method was
used to estimate the expected option life.
AZITRA,
INC.
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
For The Nine Months Ended September 30, 2023 and 2022
The
following table summarizes information about options outstanding and exercisable at September 30, 2023:
| |
|
|
|
|
Options
Outstanding |
|
|
|
Options
Exercisable |
|
| |
Exercise
Price |
|
|
|
Number
of Options at 9/30/2023 |
|
|
|
Weight
Average Remaining Contractual Life |
|
|
|
Weight
Average Exercise Price |
|
|
|
Number
of Options at 9/30/2023 |
|
|
|
Weighted
Average Remaining Contractual Life |
|
|
|
Weighted
Average Exercise Price |
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| $ |
0.48 |
|
|
|
246,137 |
|
|
|
2.3
years |
|
|
$ |
0.48 |
|
|
|
246,137 |
|
|
|
2.3
years |
|
|
$ |
0.48 |
|
| $ |
0.93 |
|
|
|
202,040 |
|
|
|
2.3
years |
|
|
$ |
0.93 |
|
|
|
201,593 |
|
|
|
2.3
year |
|
|
$ |
0.93 |
|
| $ |
1.70 |
|
|
|
800,078 |
|
|
|
7.5
years |
|
|
$ |
1.70 |
|
|
|
547,151 |
|
|
|
7.3
years |
|
|
$ |
1.70 |
|
| $ |
2.07 |
|
|
|
40,000 |
|
|
|
9.9
years |
|
|
$ |
2.07 |
|
|
|
1,667 |
|
|
|
9.9
years |
|
|
$ |
2.07 |
|
| |
|
|
|
|
1,288,255 |
|
|
|
|
|
|
|
|
|
|
|
996,548 |
|
|
|
|
|
|
|
|
|
Total
stock option activity for the nine months ended September 30, 2023, is summarized as follows:
| | |
Shares | | |
Weighted
Average Exercise Price | |
| Outstanding at December 31, 2022 | |
| 1,290,325 | | |
$ | 1.27 | |
| Granted | |
| 40,000 | | |
| 2.07 | |
| Exercised | |
| - | | |
| - | |
| Forfeited | |
| (42,070 | ) | |
| 0.87 | |
| Outstanding at September 30, 2023 | |
| 1,288,255 | | |
$ | 1.31 | |
There
are 242,345 shares available for future grant under the 2016 Plan and 1,960,000 available under the 2023 Plan at September 30, 2023.
11.
Fair Value Measurements
The
following tables summarize the fair values and levels within the fair value hierarchy in which the fair value measurements fall for assets
and liabilities measured on a recurring basis as of:
September
30, 2023
| Description | |
Level
1 | | |
Level
2 | | |
Level
3 | | |
Total | |
| | |
| | |
| | |
| | |
| |
| Liabilities | |
| | | |
| | | |
| | | |
| | |
| Common
stock warrants | |
$ | - | | |
$ | - | | |
$ | 60,933 | | |
$ | 60,933 | |
| Total | |
$ | - | | |
$ | - | | |
$ | 60,933 | | |
$ | 60,933 | |
December
31, 2022
| Description | |
Level
1 | | |
Level
2 | | |
Level
3 | | |
Total | |
| | |
| | |
| | |
| | |
| |
| Liabilities | |
| | | |
| | | |
| | | |
| | |
| Common stock
warrants | |
$ | - | | |
$ | - | | |
$ | 70,283 | | |
$ | 70,283 | |
| 2022
Convertible Notes | |
$ | - | | |
$ | - | | |
$ | 5,600,000 | | |
$ | 5,600,000 | |
| Total | |
$ | - | | |
$ | - | | |
$ | 5,670,283 | | |
$ | 5,670,283 | |
AZITRA,
INC.
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
For The Nine Months Ended September 30, 2023 and 2022
The
following table presents the changes in Level 3 instruments measured on a recurring basis for the period ended September 30, 2023:
| Balance at December 31, 2022 | |
$ | 5,670,283 | |
| Changes in fair value of
warrants | |
| (5,621 | ) |
| Changes
in fair value of 2022 Convertible Notes | |
| 800,000 | |
| Balance at March 31, 2023 | |
$ | 6,464,662 | |
| Changes in fair value of
warrants | |
| 94,036 | |
| Changes in fair value of
2022 Convertible Notes | |
| 2,830,100 | |
| Conversion
of 2022 Convertible Notes | |
| (9,230,100 | ) |
| Balance at June 30, 2023 | |
$ | 158,698 | |
| Changes
in fair value of warrants | |
| (97,765 | ) |
| Balance at September 30, 2023 | |
$ | 60,933 | |
At
September 30, 2023 and December 31, 2022, the Company estimated the fair value of the warrants using the Black-Scholes option pricing
model with the following assumptions:
| | |
September
30, 2023 | | |
December
31, 2022 | |
| Underlying common stock value | |
$ | 1.50 | | |
$ | 12.09 | |
| Expected term (years) | |
| 4.54 | | |
| 5 | |
| Expected volatility | |
| 93 | % | |
| 86 | % |
| Risk free interest rate | |
| 3 | % | |
| 3 | % |
| Dividend yield | |
| - | % | |
| - | % |
Fluctuation
in the fair value of the Company’s Common stock is the primary driver for the change in the Common Stock Warrant liability valuation
during each year. As the fair value of the Common stock increases the value to the holder of the instrument generally increases.
Prior
to their redemption, fluctuations in the various inputs, including the enterprise value, time to liquidity, volatility, and discount
rate are the primary drivers for the changes in valuation of the 2022 Convertible Notes each reporting period. As the fair value of the
enterprise value, estimated time to liquidity, volatility, and discount rate increase, the value to the holder of the 2022 Convertible
Notes generally increases.
12.
Net Loss Per Share
Basic
and diluted net loss per share were calculated as follows:
The
numerator for basic and diluted net loss per share is as follows for the nine months ended September 30, 2023 and 2022:
| | |
Nine
Months Ended | |
| | |
2023 | | |
2022 | |
| Net loss | |
$ | (8,829,996 | ) | |
$ | (6,633,790 | ) |
| Dividends on preferred
stock | |
| (1,355,347 | ) | |
| (2,076,737 | ) |
| Net loss attributable to common stockholders | |
$ | (10,185,343 | ) | |
$ | (8,710,527 | ) |
AZITRA,
INC.
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
For The Nine Months Ended September 30, 2023 and 2022
The
denominator is as follows for the nine months ended September 30, 2023 and 2022:
| | |
Nine
Months Ended | |
| | |
2023 | | |
2022 | |
| Weighted average common stock outstanding,
basic and diluted | |
| 5,173,925 | | |
| 1,043,913 | |
| $0.01 warrants | |
| 7,182 | | |
| 11,467 | |
| Total | |
| 5,181,107 | | |
| 1,055,380 | |
Net
loss per share, basic and diluted is as follows for the nine months ended September 30, 2023 and 2022:
| | |
Nine
Months Ended | |
| | |
2023 | | |
2022 | |
| Net Loss per share, basic and diluted | |
$ | (1.97 | ) | |
$ | (8.25 | ) |
The
following potential common stock equivalents, presented based on amounts outstanding at each period end, were excluded from the calculation
of diluted net loss per share for the periods indicated because including them would have had an anti-dilutive effect:
| | |
September
30, | |
| | |
2023 | | |
2022 | |
| Options to purchase shares of common
stock | |
| 1,288,255 | | |
| 1,290,318 | |
| Warrants outstanding | |
| 323,736 | | |
| 11,467 | |
| Total | |
| 1,611,991 | | |
| 1,301,785 | |
13.
Commitments and Contingencies
Legal
The
Company is subject to legal proceedings or claims which arise in the ordinary course of its business. Although occasional adverse decisions
or settlements may occur, the Company believes that the final disposition of such matters should not have a material adverse effect on
its financial position, results of operations or liquidity.
License
Agreement
Effective
January 26, 2022, the Company entered into an Exclusive License Agreement (the License Agreement) with an unrelated third party. Under
the License Agreement, the Company is granted an exclusive license for certain patents and a non-exclusive license for certain know-how.
The License Agreement continues until the later of the expiration of the last to expire licensed patent or ten years after the first
commercial sale of the first licensed therapeutic or non-therapeutic product. The Company may terminate the License Agreement at any
time by providing at least 30 days written notice to the third party. The License Agreement is also terminated upon breach of a material
obligation under the agreement or bankruptcy. Upon any termination of the License Agreement, neither party is relieved of obligations
incurred prior to the termination.
During
the nine months ended September 30, 2023 and 2022, the Company capitalized payments made under this license agreement in the amount of
$0 and $15,263, respectively. During the nine months ended September 30, 2023, the Company expensed the remainder of the capitalized
balance.
AZITRA,
INC.
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
For The Nine Months Ended September 30, 2023 and 2022
Operating
Leases
The
Company leases office and lab space in Branford, CT; Groton, CT; and Laval, Quebec. The Company’s leases expire at various dates
through May 31, 2027. Most leases are for a fixed term and for a fixed amount. The Company is not a party to any leases that have step
rent provisions, escalation clauses, capital improvement funding or payment increases based on any index or rate.
During
2020, the Company entered into a new lease agreement for the Company’s primary office and laboratory space in Branford, CT. The
Branford lease requires monthly payments of $13,033 for the first year of the lease, which increases approximately 2% in each of the
following years. The Branford lease also requires the Company to pay a pro-rata share of common area maintenance.
During
May 2021, the Company entered into a new lease for office and laboratory space in Groton, CT. The Groton lease required monthly payments
of $4,234, which was increased to $6,824 in September 2021 upon leasing additional space. The Groton lease is initially for a one-year
term, with up to three additional years renewal available.
Future
minimum payments under non-cancelable operating leases with initial or remaining terms in excess of one year during each of the next
five years follow:
| 2023 | |
$ | 81,354 | |
| 2024 | |
| 337,708 | |
| 2025 | |
| 278,928 | |
| 2026 | |
| 205,877 | |
| 2027 | |
| 73,725 | |
| Thereafter | |
| - | |
| Total future undiscounted lease payments | |
| 977,592 | |
| Less interest | |
| (62,597 | ) |
| Present value of minimum lease payments | |
$ | 914,995 | |
Rent
expense for all operating leases was $254,142 for the nine months ended September 30, 2023. The weighted average lease term for all operating
leases is 3.2 years. The weighted average discount rate for all operating leases is 4.25%.
Finance
Leases
During
2023, the Company entered into an agreement with Hewlett Packard to lease equipment. The lease requires monthly payments of $1,478, including
tax. The lease is for a 3-year term with option of purchase or extension at term end. The remaining lease term is 2.8 years and the discount
rate is 9.60%.
The
following is a schedule showing the future minimum lease payments under finance leases by years and the present value of the minimum
payments as of September 30, 2023. value of the minimum payments as of September 30, 2023.
| 2023 | |
$ | 4,435 | |
| 2024 | |
| 17,740 | |
| 2025 | |
| 17,740 | |
| 2026 | |
| 10,349 | |
| Thereafter | |
| - | |
| Total future undiscounted lease payments | |
| 50,264 | |
| Less interest | |
| (6,058 | ) |
| Present value of minimum lease payments | |
$ | 44,206 | |
Lease
expense for the finance lease was $2,580 for the nine months ended September 30, 2023. Interest expense for the finance lease was $710
for the nine months ended September 30, 2023.
AZITRA,
INC.
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
For The Nine Months Ended September 30, 2023 and 2022
14.
Retirement Plan
Effective
January 1, 2019, the Company sponsors a 401(k) plan that covers substantially all employees. In order to be eligible to participate,
an employee must complete two consecutive months of service and work a minimum of two hundred and fifty hours or work 1,000 hours in
their first year of service. Employees may make pre-tax deferrals upon meeting the Plan eligibility requirements. Effective January 1,
2020, the Plan was transitioned to a safe harbor plan in which highly compensated employees are not eligible for matching contributions
and non-highly compensated employees earn 100% match on first 3% contributed and 50% on the next 2% contributed. Total employer matching
contributions were $7,634 for the nine months ended September 30, 2023. Total employer matching contributions were $18,945 for the nine
months ended September 30, 2022.
15.
Concentration of Credit Risk
Financial
instruments that potentially subject the Company to credit risk consist principally of cash and accounts receivable.
For
the nine months ended September 30, 2023 and 2022, all service revenue was from one customer. For the nine months ended September 30,
2023, there was no grant revenue and for the nine months ended September 30, 2022, all grant revenue was from one grantor.
The
cash balance identified in the balance sheet is held in an account with a financial institution and insured by the Federal Deposit Insurance
Corporation (FDIC) up to $250,000. At times, cash maintained on deposit may be in excess of FDIC limits.
16.
Related Parties
Total
related party revenue was $596,000 for the nine months ended September 30, 2023. Total related party revenue was $253,500 for the nine
months ended September 30, 2022. Accounts receivable due from the related party was $0 and $175,000 at September 30, 2023 and December
31, 2022, respectively. Contract liabilities from the related party was $0 and $156,000 at September 30, 2023 and December 31, 2022,
respectively.
In
September 2022 the Company entered into a convertible promissory note totaling $4,350,000 of which $4,000,000 was attributable to an
entity who was also an investor in the Company’s Series A, A-1, and B Preferred Stock financing (See Note 7). This entity received
1,697,490 shares of common stock upon conversion of the promissory notes for principal and interest of $4,243,726.
17.
Subsequent Events
The
Company has evaluated events subsequent to the balance sheet date through November 14, 2023, the date these condensed financial statements
are issued.
Up
to 3,000,000 Shares of Common Stock
Up
to 3,000,000 Pre-Funded Warrants to purchase up to
3,000,000 Shares of Common Stock
PRELIMINARY
PROSPECTUS
ThinkEquity
,
2024
PART
II - INFORMATION NOT REQUIRED IN PROSPECTUS
Item
13. Other Expenses of Issuance and Distribution.
The
following table sets forth the various expenses to be incurred in connection with the sale and distribution of our common stock being
registered hereby, all of which will be borne by us (except any underwriting discounts and commissions and expenses incurred for brokerage,
accounting, tax or legal services or any other expenses incurred in disposing of the shares). All amounts are estimated except the SEC
registration fee and the FINRA filing fee.
| Description | |
Amount | |
| SEC
Registration Fee | |
$ | 1,097 | |
| FINRA Filing
Fee | |
| 2,750 | |
| Non-Accountable
Expenses to Underwriters | |
| 150,000 | |
| Printing
Expenses | |
| 25,000 | |
| Accounting
Fees and Expenses | |
| 50,000 | |
| Legal Fees
and Expenses | |
| 250,000 | |
| Transfer
Agent’s and Registrar’s Fees and Expenses | |
| 5,000 | |
| Miscellaneous
Fees | |
| 16,153 | |
| Total | |
$ | 500,000 | |
* To be provided by amendment.
Item
14. Indemnification of Directors and Officers.
The
following summary is qualified in its entirety by reference to the complete text of any statutes referred to below and the Second Amended
and Restated Certificate of Incorporation, or Certificate of Incorporation, of Azitra, Inc., a Delaware corporation.
Section
145 of the General Corporation Law of the State of Delaware (the “DGCL”) permits a Delaware corporation to indemnify any
person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding,
whether civil, criminal, administrative or investigative (other than an action by or in the right of the corporation) by reason of the
fact that the person is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the
corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise,
against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred
by the person in connection with such action, suit or proceeding if the person acted in good faith and in a manner the person reasonably
believed to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceeding, had
no reasonable cause to believe the person’s conduct was unlawful.
In
the case of an action by or in the right of the corporation, Section 145 of the DGCL permits a Delaware corporation to indemnify any
person who was or is a party or is threatened to be made a party to any threatened, pending or completed action or suit by reason of
the fact that the person is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of
the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise,
against expenses (including attorneys’ fees) actually and reasonably incurred by the person in connection with such action, suit
or proceeding if the person acted in good faith and in a manner the person reasonably believed to be in or not opposed to the best interests
of the corporation and except that no indemnification shall be made in respect of any claim, issue or matter as to which such person
shall have been adjudged to be liable to the corporation unless and only to the extent that the Court of Chancery or the court in which
such action or suit was brought shall determine upon application that, despite the adjudication of liability but in view of all the circumstances
of the case, such person is fairly and reasonably entitled to indemnity for such expenses that the Court of Chancery or such other court
shall deem proper.
Section
145 of the DGCL also permits a Delaware corporation to purchase and maintain insurance on behalf of any person who is or was a director,
officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee
or agent of another corporation, partnership, joint venture, trust or other enterprise against any liability asserted against such person
and incurred by such person in any such capacity, or arising out of such person’s status as such, whether or not the corporation
would have the power to indemnify such person against such liability under Section 145 of the DGCL.
Our
Certificate of Incorporation states that to the fullest extent permitted by the DGCL our directors shall not be personally liable to
us or to our stockholders for monetary damages for breach of fiduciary duty as a director. If the DGCL is amended after the date hereof
to authorize corporate action further eliminating or limiting the personal liability of directors, then the liability of our directors
shall be eliminated or limited to the fullest extent permitted by the DGCL, as so amended.
Our
Certificate of Incorporation requires us, to the fullest extent permitted by applicable law, to provide indemnification of (and advancement
of expenses to) our directors and officers, and authorizes us, to the fullest extent permitted by applicable law, to provide indemnification
of (and advancement of expenses to) to other employees and agents (and any other persons to which the DGCL permits us to provide indemnification)
through bylaw provisions, agreements with such directors, officers, employees, agents or other persons, vote of stockholders or disinterested
directors or otherwise, subject only to limits created by the DGCL with respect to actions for breach of duty to our corporation, our
stockholders and others.
Our
Certificate of Incorporation provides that we shall, to the maximum extent and in the manner permitted by the DGCL, indemnify each of
our directors, officers and all other persons we have the power to indemnify under Section 145 of the DGCL against expenses (including
attorneys’ fees), judgments, fines, settlements and other amounts actually and reasonably incurred in connection with any proceeding,
arising by reason of the fact that such person is or was a director of the Company. We may maintain insurance, at our expense, to protect
the Company and any of our directors, officers, employees or agents against any such expense, liability or loss, whether or not we have
the power to indemnify such person.
We
have entered into indemnification agreements with each of our directors and executive officers that may be broader than the specific
indemnification provisions contained in the DGCL. These indemnification agreements require us, among other things, to indemnify our directors
and executive officers against liabilities that may arise by reason of their status or service. These indemnification agreements also
require us to advance all expenses incurred by the directors and executive officers in investigating or defending any such action, suit,
or proceeding. We believe that these agreements are necessary to attract and retain qualified individuals to serve as directors and executive
officers.
Prior
to the closing of this offering, we plan to enter into an underwriting agreement, which will provide that the underwriters are obligated,
under some circumstances, to indemnify our directors, officers and controlling persons against specified liabilities.
Item
15. Recent Sales of Unregistered Securities.
Issuances
of capital stock
The
following list sets forth information regarding all unregistered securities sold by us over the three-year period preceding the date
of the prospectus that forms a part of this registration statement.
In
January 2022, we sold to one investor an unsecured convertible promissory note in the principal amount of $1,000,000. In January 2023,
the principal amount of the note, along with all accrued interest, was converted into 23,432 shares of our Series B convertible preferred
stock. Subsequently, these shares were converted into common stock upon the consummation of the IPO.
In
September 2022, we conducted the placement of our unsecured convertible promissory notes in the aggregate principal amount of $4.35 million
to five investors. The principal amount of the notes, along with all accrued and unpaid interest thereunder, converted into 1,846,020
shares of our common stock upon the consummation of the IPO.
No
underwriters were involved in the foregoing issuances of securities. We believe the offers, sales and issuances of the above securities
by us were exempt from registration under the Securities Act by virtue of Section 4(a)(2) of the Securities Act and Rule 506 thereunder
as transactions not involving a public offering. All of the investors were accredited investors as such term is defined in Rule 501 under
the Securities Act. The recipients of the securities in each of these transactions represented their intentions to acquire the securities
for investment only and not with a view to or for sale in connection with any distribution thereof, and appropriate legends were placed
upon the stock certificates, notes and warrants issued in these transactions. All recipients had adequate access, through their relationships
with us, to information about our Company. The sales of these securities were made without any general solicitation or advertising.
Item
16. Exhibits and Financial Statement Schedules.
| Exhibit
No. |
|
Description
of Document |
|
Method
of Filing |
| |
|
|
|
|
| 1.1 |
|
Form
of Underwriting Agreement. |
|
To
be filed by amendment. |
| |
|
|
|
|
| 3.1 |
|
Second
Amended and Restated Certificate of Incorporation of the Registrant. |
|
Incorporated
herein by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K filed on June 21, 2023 (File No. 001-41705). |
| |
|
|
|
|
| 3.2 |
|
Second
Amended and Restated Bylaws of the Registrant. |
|
Incorporated
herein by reference to Exhibit 3.2 to the Company’s Current Report on Form 8-K filed on June 21, 2023 (File No. 001-41705). |
| |
|
|
|
|
| 4.1 |
|
Specimen
Certificate representing shares of Common Stock. |
|
Incorporated
herein by reference to Exhibit 4.1 to the Company’s Form S-1 filed on June 13, 2023 (File No. 333-269876). |
| |
|
|
|
|
| 4.2 |
|
Form
of Warrant issued to private placement investors. |
|
Incorporated
herein by reference to Exhibit 4.2 to the Company’s Form S-1 filed on June 13, 2023 (File No. 333-269876). |
| |
|
|
|
|
| 4.3 |
|
Form
of Representative’s Warrant dated June 20, 2023 issued to ThinkEquity LLC. |
|
Incorporated
herein by reference to Exhibit 4.3 to the Company’s Form S-1 filed on June 13, 2023 (File No. 333-269876). |
| |
|
|
|
|
| 4.4 |
|
Form of Pre-Funded Common Stock Purchase Warrant |
|
Filed electronically herewith. |
| |
|
|
|
|
| 5.1 |
|
Opinion
of Greenberg Traurig, LLP with respect to the common stock. |
|
To
be filed by amendment. |
| |
|
|
|
|
| 5.2 |
|
Opinion of Greenberg Traurig, LLP with respect to the Pre-Funded Warrants. |
|
To be filed by amendment. |
| |
|
|
|
|
| 10.1+ |
|
Azitra,
Inc. 2016 Stock Incentive Plan. |
|
Incorporated
herein by reference to Exhibit 10.2 to the Company’s Form S-1 filed on June 13, 2023 (File No. 333-269876). |
| 10.2+ |
|
Azitra,
Inc. 2023 Stock Incentive Plan. |
|
Incorporated
herein by reference to Exhibit 10.5 to the Company’s Form S-1 filed on June 13, 2023 (File No. 333-269876). |
| 10.3+ |
|
Executive
Employment Agreement dated April 22, 2021 between the Registrant and Francisco D. Salva. |
|
Incorporated
herein by reference to Exhibit 10.4 to the Company’s Form S-1 filed on June 13, 2023 (File No. 333-269876). |
+
Indicates management compensatory plan, contract or arrangement.
(b)
Financial Statement Schedules.
All
schedules have been omitted because they are not required or are not applicable.
Item
17. Undertakings.
The
undersigned registrant hereby undertakes to provide to the underwriter at the closing specified in the underwriting agreements, certificates
in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser.
Insofar
as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons
of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities
and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable.
In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred
or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is
asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless
in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the
question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final
adjudication of such issue.
The
undersigned registrant undertakes that:
(1)
For purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus as filed as part
of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule
424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared
effective.
(2)
For the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus
shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at
that time shall be deemed to be the initial bona fide offering thereof.
SIGNATURES
Pursuant
to the requirements of the Securities Act of 1933, the registrant has duly caused this registration statement to be signed on its behalf
by the undersigned, thereunto duly authorized, in the City of Branford, State of Connecticut, on January 19, 2024.
|
|
| |
AZITRA,
INC. |
| |
|
| |
/s/
Francisco D. Salva |
| |
Francisco
D. Salva |
| |
Chief
Executive Officer and Director |
POWER
OF ATTORNEY
Each
person whose signature appears below constitutes and appoints Francisco D. Salva, his true and lawful attorney-in-fact and agent, with
full power of substitution and resubstitution, for him and in his name, place and stead, in any and all capacities, to sign any and all
amendments (including post-effective amendments) to this registration statement, any subsequent registration statements pursuant to Rule
462 of the Securities Act of 1933, as amended, and to file the same, with all exhibits thereto and other documents in connection therewith,
with the Securities and Exchange Commission, granting unto said attorney-in-fact and agent, full power and authority to do and perform
each and every act and thing requisite and necessary to be done in and about the premises, as fully to all intents and purposes as he
might or could do in person, hereby ratifying and confirming all that said attorney-in-fact and agent, or his substitute or substitutes,
may lawfully do or cause to be done by virtue hereof. This power of attorney may be executed in counterparts.
Pursuant
to the requirements of the Securities Act of 1933, this registration statement has been signed by the following persons in the capacities
and on the dates indicated.
Signature |
|
Title |
|
Date |
| |
|
|
|
|
/s/ Francisco D.
Salva |
|
President, |
|
|
|
Francisco
D. Salva |
|
Chief
Executive Officer and Director
(Principal
Executive Officer) |
|
January
19, 2024 |
| |
|
|
|
|
/s/
Norman Staskey |
|
Chief
Financial Officer, |
|
|
|
Norman
Staskey |
|
Treasurer
and Secretary
(Principal
Financial and Accounting Officer) |
|
January
19, 2024 |
| |
|
|
|
|
/s/
Travis Whitfill |
|
|
|
|
|
Travis
Whitfill |
|
Chief
Operating Officer and Director |
|
January
19, 2024 |
| |
|
|
|
|
/s/
Andrew McClary |
|
|
|
|
|
Andrew
McClary |
|
Director |
|
January
19, 2024 |
| |
|
|
|
|
/s/
Barbara Ryan |
|
|
|
|
|
Barbara
Ryan |
|
Director |
|
January
19, 2024 |
| |
|
|
|
|
/s/
John Schroer |
|
|
|
|
|
John
Schroer |
|
Director |
|
January
19, 2024 |
Exhibit
4.4
PRE-FUNDED
COMMON STOCK PURCHASE WARRANT
Azitra,
Inc.
Warrant
Shares: _______
Issue
Date: January ___, 2024
THIS
PRE-FUNDED COMMON STOCK PURCHASE WARRANT (the “Warrant”) certifies that, for value received, _____________ or its
assigns (the “Holder”) is entitled, upon the terms and subject to the limitations on exercise and the conditions hereinafter
set forth, at any time on or after the Issue Date and until this Warrant is exercised in full (the “Termination Date”)
but not thereafter, to subscribe for and purchase from Azitra, Inc., a Delaware corporation (the “Company”), up to
______ shares of Common Stock (as subject to adjustment hereunder, the “Warrant Shares”). The purchase price of one
share of Common Stock under this Warrant shall be equal to the Exercise Price, as defined in Section 2(b).
Definitions.
In addition to the terms defined elsewhere in this Warrant, the following terms have the meanings indicated in this Section
1:
“Affiliate”
means any Person that, directly or indirectly through one or more intermediaries, controls or is controlled by or is under common control
with a Person, as such terms are used in and construed under Rule 405 under the Securities Act.
“Commission”
means the United States Securities and Exchange Commission.
“Common
Stock” means the common stock of the Company, par value $0.0001 per share, and any other class of securities into which such
securities may hereafter be reclassified or changed.
“Common
Stock Equivalents” means any securities of the Company or the Subsidiaries which would entitle the holder thereof to acquire
at any time Common Stock, including, without limitation, any debt, preferred stock, right, option, warrant or other instrument that is
at any time convertible into or exercisable or exchangeable for, or otherwise entitles the holder thereof to receive, Common Stock.
“Exchange
Act” means the Securities Exchange Act of 1934, as amended, and the rules and regulations promulgated thereunder.
“Liens”
means a lien, charge pledge, security interest, encumbrance, right of first refusal, preemptive right or other restriction.
“Person”
means an individual or corporation, partnership, trust, incorporated or unincorporated association, joint venture, limited liability
company, joint stock company, government (or an agency or subdivision thereof) or other entity of any kind.
“Proceeding”
means an action, claim, suit, investigation or proceeding (including, without limitation, an informal investigation or partial proceeding,
such as a deposition), whether commenced or threatened.
“Registration
Statement” means the Company’s registration statement on Form S-1 (File No. 333- ).
“Rule
144” means Rule 144 promulgated by the Commission pursuant to the Securities Act, as such Rule may be amended or interpreted
from time to time, or any similar rule or regulation hereafter adopted by the Commission having substantially the same purpose and effect
as such Rule.
“Securities
Act” means the Securities Act of 1933, as amended, and the rules and regulations promulgated thereunder.
“Trading
Day” means a day on which the Common Stock is traded on a Trading Market.
“Trading
Market” means any of the following markets or exchanges on which the Common Stock is listed or quoted for trading on the date
in question: the OTCQB, OTCQX or Pink Open Market operated by OTC Markets Group, the NYSE American, the Nasdaq Capital Market, the Nasdaq
Global Market, the Nasdaq Global Select Market, or the New York Stock Exchange (or any successors to any of the foregoing).
“Transfer
Agent” means VStock Transfer LLC and any successor transfer agent of the Company.
“Warrants”
means this Warrant and other Pre-Funded Common Stock Purchase Warrants issued by the Company pursuant to the Registration Statement.
Exercise.
Exercise
of the purchase rights represented by this Warrant may be made, in whole or in part, at any time or times on or after the Issue Date
and on or before the Termination Date by delivery to the Company of a duly executed facsimile copy (or e-mail attachment) of the Notice
of Exercise in the form annexed hereto (the “Notice of Exercise”). Within the earlier of (i) two (2) Trading Days
and (ii) the number of Trading Days comprising the Standard Settlement Period (as defined in Section 2(d)(i) herein) following the date
of exercise as aforesaid, the Holder shall deliver to the Company the aggregate Exercise Price for the Warrant Shares specified in the
applicable Notice of Exercise by wire transfer or cashier’s check drawn on a United States bank unless the cashless exercise procedure
specified in Section 2(c) below is specified in the applicable Notice of Exercise. No ink-original Notice of Exercise shall be required,
nor shall any medallion guarantee (or other type of guarantee or notarization) of any Notice of Exercise form be required. Notwithstanding
anything herein to the contrary, the Holder shall not be required to physically surrender this Warrant to the Company until the Holder
has purchased all of the Warrant Shares available hereunder and the Warrant has been exercised in full, in which case, the Holder shall
surrender this Warrant to the Company for cancellation within three (3) Trading Days of the date on which the final Notice of Exercise
is delivered to the Company. Partial exercises of this Warrant resulting in purchases of a portion of the total number of Warrant Shares
available hereunder shall have the effect of lowering the outstanding number of Warrant Shares purchasable hereunder in an amount equal
to the applicable number of Warrant Shares purchased in connection with such partial exercise. The Holder and the Company shall maintain
records showing the number of Warrant Shares purchased and the date of such purchases. The Company shall deliver any objection to any
Notice of Exercise within one (1) Trading Day of receipt of such notice. The Holder and any assignee, by acceptance of this Warrant,
acknowledge and agree that, by reason of the provisions of this paragraph, following the purchase of a portion of the Warrant Shares
hereunder, the number of Warrant Shares available for purchase hereunder at any given time may be less than the amount stated on the
face hereof.
Exercise
Price. The aggregate exercise price of this Warrant, except for a nominal exercise price of $0.001 per Warrant Share, was
pre-funded to the Company on or prior to the Issue Date and, consequently, no additional consideration (other than the nominal exercise
price of $0.001 per Warrant Share) shall be required to be paid by the Holder to any Person to effect any exercise of this Warrant.
The Holder shall not be entitled to the return or refund of all, or any portion, of such pre-paid aggregate exercise price under any
circumstance or for any reason whatsoever, including in the event this Warrant shall not have been exercised prior to the Termination
Date. The remaining unpaid exercise price per share of Common Stock under this Warrant shall be $0.001, subject to adjustment
hereunder (the “Exercise Price”).
Cashless
Exercise. This Warrant may also be exercised, in whole or in part, at such time by means of a “cashless exercise” in
which the Holder shall be entitled to receive a number of Warrant Shares for the deemed surrender of the Warrant in whole or in part
equal to the quotient obtained by dividing [(A-B) (X)] by (A), where:
| |
(A)
= |
as
applicable: (i) the Closing Sale Price of the Common Stock on the Trading Day immediately preceding the date of the applicable Notice
of Exercise if such Notice of Exercise is (1) both executed and delivered pursuant to Section 2(a) hereof on a day that is not a
Trading Day or (2) both executed and delivered pursuant to Section 2(a) hereof on a Trading Day prior to the opening of “regular
trading hours” (as defined in Rule 600(b)(68) of Regulation NMS promulgated under the federal securities laws) on such Trading
Day, (ii) at the option of the Holder, either (x) the VWAP on the Trading Day immediately preceding the date of the applicable Notice
of Exercise or (y) the Bid Price of the Common Stock as of the time of the Holder’s execution of the applicable Notice of Exercise
if such Notice of Exercise is executed during “regular trading hours” on a Trading Day and is delivered within two (2)
hours thereafter pursuant to Section 2(a) hereof (including until two (2) hours after the close of “regular trading hours”
on a Trading Day), or (iii) the Closing Sale Price of the Common Stock on the date of the applicable Notice of Exercise if the date
of such Notice of Exercise is a Trading Day and such Notice of Exercise is both executed and delivered pursuant to Section 2(a) hereof
after the close of “regular trading hours” on such Trading Day; |
| |
|
|
| |
(B)
= |
the
Exercise Price of this Warrant, as adjusted hereunder; and |
| |
|
|
| |
(X)
= |
the
number of Warrant Shares that would be issuable upon exercise of this Warrant in accordance with the terms of this Warrant if such
exercise were by means of a cash exercise rather than a cashless exercise. |
The
issue price for each such Warrant Share to be issued pursuant to the cashless exercise of a Warrant will be equal to (B), as defined
above, and the total issue price for the aggregate number of Warrant Shares issued pursuant to the cashless exercise of a Warrant will
be deemed paid and satisfied in full by the deemed surrender to the Company of the portion of such Warrant being exercised in accordance
with this Section 1(c). Notwithstanding anything herein to the contrary, the Company shall not be required to make any cash payments
or net cash settlement to the Holder in lieu of delivery of the Warrant Shares. If Warrant Shares are issued in such a cashless exercise,
the parties acknowledge and agree that in accordance with Section 3(a)(9) of the Securities Act, the Warrant Shares shall take on the
registered characteristics of the Warrants being exercised. The Company agrees not to take any position contrary to this Section 2(c).
“Bid
Price” means, for any security as of the particular time of determination, the bid price for such security on the Trading Market
as reported by Bloomberg as of such time of determination, or, if the Trading Market is not the principal securities exchange or trading
market for such security, the bid price of such security on the principal securities exchange or trading market where such security is
listed or traded as reported by Bloomberg as of such time of determination, or if the foregoing does not apply, the bid price of such
security in the over-the-counter market on the electronic bulletin board for such security as reported by Bloomberg as of such time of
determination, or, if no bid price is reported for such security by Bloomberg as of such time of determination, the average of the bid
prices of any market makers for such security as reported on the Pink Open Market as of such time of determination. If the Bid Price
cannot be calculated for a security as of the particular time of determination on any of the foregoing bases, the Bid Price of such security
as of such time of determination shall be the fair market value as mutually determined by the Company and the Holder. If the Company
and the Holder are unable to agree upon the fair market value of such security, then such fair market value shall be determined pursuant
to the provisions set forth in clause (d) of the definition of VWAP. All such determinations to be appropriately adjusted for any stock
dividend, share split, share consolidation, reclassification or other similar transaction during the applicable calculation period.
“Closing
Sale Price” means, for any security as of any date, the last closing trade price for such security on the Trading Market, as
reported by Bloomberg, or, if the Trading Market begins to operate on an extended hours basis and does not designate the closing trade
price, then the last trade price of such security prior to 4:00:00 p.m., New York time, as reported by Bloomberg, or, if the Trading
Market is not the principal securities exchange or trading market for such security, the last trade price of such security on the principal
securities exchange or trading market where such security is listed or traded as reported by Bloomberg, or if the foregoing do not apply,
the last trade price of such security in the over-the-counter market on the electronic bulletin board for such security as reported by
Bloomberg, or, if no last trade price is reported for such security by Bloomberg, the average of the ask prices of any market makers
for such security as reported on the in the OTC Link or on the Pink Open Market. If the Closing Sale Price cannot be calculated for a
security on a particular date on any of the foregoing bases, Closing Sale Price of such security on such date shall be the fair market
value as mutually determined by the Company and the Holder. If the Company and the Holder are unable to agree upon the fair market value
of such security, then such fair market value shall be determined pursuant to the provisions set forth in clause (d) of the definition
of VWAP. All such determinations to be appropriately adjusted for any stock dividend, share split, share consolidation, reclassification
or other similar transaction during the applicable calculation period.
“VWAP”
means, for any date, the price determined by the first of the following clauses that applies: (a) if the Common Stock is then listed
or quoted for trading on a Trading Market other than the OTCQB, OTCQX or Pink Open Market operated by OTC Markets Group, the daily volume
weighted average price of the Common Stock for such date (or the nearest preceding date) on the Trading Market on which the Common Stock
is then listed or quoted as reported by Bloomberg L.P. (based on a Trading Day from 9:30 a.m. (New York City time) to 4:02 p.m. (New
York City time)), (b) if the Common Stock is then quoted for trading on the OTCQB or OTCQX operated by OTC Markets Group, the volume
weighted average price of the Common Stock for such date (or the nearest preceding date) on OTCQB or OTCQX as applicable, or (c) in all
other cases, the fair market value of a share of Common Stock as determined by an independent appraiser selected in good faith by the
holders of a majority in interest of the Warrants then outstanding and reasonably acceptable to the Company, the fees and expenses of
which shall be paid by the Company.
Mechanics
of Exercise.
Delivery
of Warrant Shares Upon Exercise. The Company shall cause the Warrant Shares purchased hereunder to be transmitted by the Transfer
Agent to the Holder by crediting the account of the Holder’s or its designee’s balance account with The Depository Trust
Company through its Deposit or Withdrawal at Custodian system (the “DWAC”) if the Company is then a participant in
such system and either (A) there is an effective registration statement permitting the issuance of the Warrant Shares to or resale of
the Warrant Shares by the Holder or (B) this Warrant is being exercised via cashless exercise, and otherwise by physical delivery of
a certificate, registered in the Company’s share register in the name of the Holder or its designee, for the number of Warrant
Shares to which the Holder is entitled pursuant to such exercise to the address specified by the Holder in the Notice of Exercise by
the date that is the earlier of (i) two (2) Trading Days and (ii) the number of Trading Days comprising the Standard Settlement Period
after the delivery to the Company of the Notice of Exercise (such date, the “Warrant Share Delivery Date”). Upon delivery
of the Notice of Exercise, the Holder shall be deemed for all corporate purposes to have become the Holder of record of the Warrant Shares
with respect to which this Warrant has been exercised, irrespective of the date of delivery of the Warrant Shares, provided that payment
of the aggregate Exercise Price (other than in the case of a cashless exercise) is received within the earlier of (i) two (2) Trading
Days after the delivery to the Company of the Notice of Exercise and (ii) the number of Trading Days comprising the Standard Settlement
Period following delivery of the Notice of Exercise. If the Company fails for any reason to deliver to the Holder the Warrant Shares
subject to a Notice of Exercise by the Warrant Share Delivery Date, the Company shall pay to the Holder, in cash, as liquidated damages
and not as a penalty, for each $1,000 of Warrant Shares subject to such exercise (based on the VWAP of the Common Stock on the date of
the applicable Notice of Exercise), $10 per Trading Day (increasing to $20 per Trading Day on the third Trading Day after the Warrant
Share Delivery Date) for each Trading Day after such Warrant Share Delivery Date until such Warrant Shares are delivered to said Holder
or the Holder rescinds such exercise. The Company agrees to maintain a Transfer Agent that is a participant in the Fast Automated Securities
Transfer or FAST program so long as this Warrant remains outstanding and exercisable. As used herein, “Standard Settlement Period”
means the standard settlement period, expressed in a number of Trading Days, on the Company’s primary Trading Market with respect
to the Common Stock as in effect on the date of delivery of the Notice of Exercise. Notwithstanding the foregoing, with respect to any
Notice(s) of Exercise delivered on or prior to 12:00 p.m. (New York City time) on the Issue Date, which may be delivered at any time
after the time of execution of the Underwriting Agreement, dated January [●], 2024 between the Company and ThinkEquity LLC, the
Company agrees to deliver the Warrant Shares subject to such notice(s) by 4:00 p.m. (New York City time) on the Issue Date.
Delivery
of New Warrants Upon Exercise. If this Warrant shall have been exercised in part, the Company shall, at the request of a Holder and
upon surrender of this Warrant certificate, at the time of delivery of the Warrant Shares, deliver to the Holder a new Warrant evidencing
the rights of the Holder to purchase the unpurchased Warrant Shares called for by this Warrant, which new Warrant shall in all other
respects be identical with this Warrant.
Rescission
Rights. If the Company fails to cause the Transfer Agent to transmit to the Holder the Warrant Shares pursuant to Section 2(d)(i)
by the Warrant Share Delivery Date, then the Holder will have the right to rescind such exercise.
Compensation
for Buy-In on Failure to Timely Deliver Warrant Shares Upon Exercise. In addition to any other rights available to the Holder, if
the Company fails to cause the Transfer Agent to transmit to the Holder the Warrant Shares in accordance with the provisions of Section
2(d)(i) above pursuant to an exercise on or before the Warrant Share Delivery Date, and if after such date the Holder is required by
its broker to purchase (in an open market transaction or otherwise) or the Holder’s brokerage firm otherwise purchases, shares
of Common Stock to deliver in satisfaction of a sale by the Holder of the Warrant Shares which the Holder anticipated receiving upon
such exercise (a “Buy-In”), then the Company shall (A) pay in cash to the Holder the amount, if any, by which (x)
the Holder’s total purchase price (including brokerage commissions, if any) for the shares of Common Stock so purchased exceeds
(y) the amount obtained by multiplying (1) the number of Warrant Shares that the Company was required to deliver to the Holder in connection
with the exercise at issue by (2) the price at which the sell order giving rise to such purchase obligation was executed, and (B) at
the option of the Holder, either reinstate the portion of the Warrant and equivalent number of Warrant Shares for which such exercise
was not honored (in which case such exercise shall be deemed rescinded) or deliver to the Holder the number of shares of Common Stock
that would have been issued had the Company timely complied with its exercise and delivery obligations hereunder. For example, if the
Holder purchases shares of Common Stock having a total purchase price of $11,000 to cover a Buy-In with respect to an attempted exercise
of Common Stock with an aggregate sale price giving rise to such purchase obligation of $10,000, under clause (A) of the immediately
preceding sentence the Company shall be required to pay the Holder $1,000. The Holder shall provide the Company written notice indicating
the amounts payable to the Holder in respect of the Buy-In and, upon request of the Company, evidence of the amount of such loss. Nothing
herein shall limit a Holder’s right to pursue any other remedies available to it hereunder, at law or in equity including, without
limitation, a decree of specific performance and/or injunctive relief with respect to the Company’s failure to timely deliver shares
of Common Stock upon exercise of the Warrant as required pursuant to the terms hereof.
No
Fractional Shares or Scrip. No fractional shares or scrip representing fractional shares shall be issued upon the exercise of this
Warrant. As to any fraction of a share which the Holder would otherwise be entitled to purchase upon such exercise, the Company shall,
at its election, either pay a cash adjustment in respect of such final fraction in an amount equal to such fraction multiplied by the
Exercise Price or round up to the next whole share.
Charges,
Taxes and Expenses. Issuance of Warrant Shares shall be made without charge to the Holder for any issue or transfer tax or other
incidental expense in respect of the issuance of such Warrant Shares, all of which taxes and expenses shall be paid by the Company, and
such Warrant Shares shall be issued in the name of the Holder or in such name or names as may be directed by the Holder; provided,
however, that in the event that Warrant Shares are to be issued in a name other than the name of the Holder, this Warrant when
surrendered for exercise shall be accompanied by the Assignment Form attached hereto duly executed by the Holder and the Company may
require, as a condition thereto, the payment of a sum sufficient to reimburse it for any transfer tax incidental thereto. The Company
shall pay all Transfer Agent fees required for same-day processing of any Notice of Exercise and all fees to the Depository Trust Company
(or another established clearing corporation performing similar functions) required for same-day electronic delivery of the Warrant Shares.
Closing
of Books. The Company shall not close its stockholder books or records in any manner which prevents the timely exercise of this Warrant,
pursuant to the terms hereof.
Holder’s
Exercise Limitations. The Company shall not effect any exercise of this Warrant, and a Holder shall not have the right to exercise
any portion of this Warrant, pursuant to Section 2 or otherwise, to the extent that after giving effect to such issuance after exercise
as set forth on the applicable Notice of Exercise, the Holder (together with the Holder’s Affiliates, and any other Persons acting
as a group together with the Holder or any of the Holder’s Affiliates (such Persons, “Attribution Parties”)),
would beneficially own in excess of the Beneficial Ownership Limitation (as defined below). For purposes of the foregoing sentence, the
number of shares of Common Stock beneficially owned by the Holder and its Affiliates and Attribution Parties shall include the number
of shares of Common Stock issuable upon exercise of this Warrant with respect to which such determination is being made, but shall exclude
the number of shares of Common Stock which would be issuable upon (i) exercise of the remaining, non-exercised portion of this Warrant
beneficially owned by the Holder or any of its Affiliates or Attribution Parties and (ii) exercise or conversion of the unexercised or
non-converted portion of any other securities of the Company (including, without limitation, any other Common Stock Equivalents) subject
to a limitation on conversion or exercise analogous to the limitation contained herein beneficially owned by the Holder or any of its
Affiliates or Attribution Parties. Except as set forth in the preceding sentence, for purposes of this Section 2(e), beneficial ownership
shall be calculated in accordance with Section 13(d) of the Exchange Act and the rules and regulations promulgated thereunder, it being
acknowledged by the Holder that the Company is not representing to the Holder that such calculation is in compliance with Section 13(d)
of the Exchange Act and the Holder is solely responsible for any schedules required to be filed in accordance therewith. To the extent
that the limitation contained in this Section 2(e) applies, the determination of whether this Warrant is exercisable (in relation to
other securities owned by the Holder together with any Affiliates and Attribution Parties) and of which portion of this Warrant is exercisable
shall be in the sole discretion of the Holder, and the submission of a Notice of Exercise shall be deemed to be the Holder’s determination
of whether this Warrant is exercisable (in relation to other securities owned by the Holder together with any Affiliates and Attribution
Parties) and of which portion of this Warrant is exercisable, in each case subject to the Beneficial Ownership Limitation, and the Company
shall have no obligation to verify or confirm the accuracy of such determination. In addition, a determination as to any group status
as contemplated above shall be determined in accordance with Section 13(d) of the Exchange Act and the rules and regulations promulgated
thereunder. For purposes of this Section 2(e), in determining the number of outstanding shares of Common Stock, a Holder may rely on
the number of outstanding shares of Common Stock as reflected in (A) the Company’s most recent periodic or annual report filed
with the Commission, as the case may be, (B) a more recent public announcement by the Company or (C) a more recent written notice by
the Company or the Transfer Agent setting forth the number of shares of Common Stock outstanding. Upon the written or oral request of
a Holder, the Company shall within two Trading Days confirm orally and in writing to the Holder the number of shares of Common Stock
then outstanding. In any case, the number of outstanding shares of Common Stock shall be determined after giving effect to the conversion
or exercise of securities of the Company, including this Warrant, by the Holder or its Affiliates or Attribution Parties since the date
as of which such number of outstanding shares of Common Stock was reported. The “Beneficial Ownership Limitation”
shall be 4.99% (or, upon election by a Holder prior to the issuance of any Warrants, 9.99%) of the number of shares of Common Stock outstanding
immediately after giving effect to the issuance of Common Stock issuable upon exercise of this Warrant. The Holder, upon notice to the
Company, may increase or decrease the Beneficial Ownership Limitation provisions of this Section 2(e), provided that the Beneficial Ownership
Limitation in no event exceeds 9.99% of the number of shares of Common Stock outstanding immediately after giving effect to the issuance
of Common Stock upon exercise of this Warrant held by the Holder and the provisions of this Section 2(e) shall continue to apply. Any
increase in the Beneficial Ownership Limitation will not be effective until the 61st day after such notice is delivered to
the Company. The provisions of this paragraph shall be construed and implemented in a manner otherwise than in strict conformity with
the terms of this Section 2(e) to correct this paragraph (or any portion hereof) which may be defective or inconsistent with the intended
Beneficial Ownership Limitation herein contained or to make changes or supplements necessary or desirable to properly give effect to
such limitation. The limitations contained in this paragraph shall apply to a successor holder of this Warrant.
Certain
Adjustments.
Stock
Dividends and Splits. If the Company, at any time while this Warrant is outstanding: (i) pays a stock dividend or otherwise makes
a distribution or distributions on shares of its Common Stock or any other equity or equity equivalent securities payable in shares of
Common Stock (which, for avoidance of doubt, shall not include any Common Stock issued by the Company upon exercise of this Warrant),
(ii) subdivides outstanding shares of Common Stock into a larger number of shares, (iii) combines (including by way of reverse stock
split or consolidation) outstanding Common Stock into a smaller number of shares, or (iv) issues by reclassification of shares of Common
Stock any shares of the Company, then in each case the Exercise Price shall be multiplied by a fraction of which the numerator shall
be the number of shares of Common Stock (excluding treasury shares, if any) outstanding immediately before such event and of which the
denominator shall be the number of shares of Common Stock outstanding immediately after such event, and the number of shares issuable
upon exercise of this Warrant shall be proportionately adjusted such that the aggregate Exercise Price of this Warrant shall remain unchanged.
Any adjustment made pursuant to this Section 3(a) shall become effective immediately after the record date for the determination of stockholders
entitled to receive such dividend or distribution and shall become effective immediately after the effective date in the case of a subdivision,
combination or re-classification.
Subsequent
Rights Offerings. In addition to any adjustments pursuant to Section 3(a) above, if at any time the Company grants, issues or sells
any Common Stock Equivalents or rights to purchase stock, warrants, securities or other property pro rata to the record holders of shares
of Common Stock (the “Purchase Rights”), then the Holder will be entitled to acquire, upon the terms applicable to
such Purchase Rights, the aggregate Purchase Rights which the Holder could have acquired if the Holder had held the number of shares
of Common Stock acquirable upon complete exercise of this Warrant (without regard to any limitations on exercise hereof, including without
limitation, the Beneficial Ownership Limitation) immediately before the date on which a record is taken for the grant, issuance or sale
of such Purchase Rights, or, if no such record is taken, the date as of which the record holders of shares of Common Stock are to be
determined for the grant, issue or sale of such Purchase Rights (provided, however, to the extent that the Holder’s right to participate
in any such Purchase Right would result in the Holder exceeding the Beneficial Ownership Limitation, then the Holder shall not be entitled
to participate in such Purchase Right to such extent (or beneficial ownership of such shares of Common Stock as a result of such Purchase
Right to such extent) and such Purchase Right to such extent shall be held in abeyance for the Holder until such time, if ever, as its
right thereto would not result in the Holder exceeding the Beneficial Ownership Limitation).
Pro
Rata Distributions. During such time as this Warrant is outstanding, if the Company shall declare or make any dividend or other distribution
of its assets (or rights to acquire its assets) to holders of shares of Common Stock, by way of return of capital or otherwise (including,
without limitation, any distribution of cash, stock or other securities, property or options by way of a dividend, spin off, reclassification,
corporate rearrangement, scheme of arrangement or other similar transaction) (a “Distribution”), at any time after
the issuance of this Warrant, then, in each such case, the Holder shall be entitled to participate in such Distribution to the same extent
that the Holder would have participated therein if the Holder had held the number of shares of Common Stock acquirable upon complete
exercise of this Warrant (without regard to any limitations on exercise hereof, including without limitation, the Beneficial Ownership
Limitation) immediately before the date of which a record is taken for such Distribution, or, if no such record is taken, the date as
of which the record holders of shares of Common Stock are to be determined for the participation in such Distribution (provided,
however, to the extent that the Holder’s right to participate in any such Distribution would result in the Holder exceeding
the Beneficial Ownership Limitation, then the Holder shall not be entitled to participate in such Distribution to such extent (or in
the beneficial ownership of any shares of Common Stock as a result of such Distribution to such extent) and the portion of such Distribution
shall be held in abeyance for the benefit of the Holder until such time, if ever, as its right thereto would not result in the Holder
exceeding the Beneficial Ownership Limitation).
Fundamental
Transaction. If, at any time while this Warrant is outstanding, (i) the Company, directly or indirectly, in one or more related transactions
effects any merger or amalgamation or consolidation of the Company with or into another Person, (ii) the Company, directly or indirectly,
effects any sale, lease, license, assignment, transfer, conveyance or other disposition of all or substantially all of its assets in
one or a series of related transactions, (iii) any, direct or indirect, purchase offer, tender offer or exchange offer (whether by the
Company or another Person) is completed pursuant to which holders of Common Stock are permitted to sell, tender or exchange their shares
for other securities, cash or property and has been accepted by the holders of 50% or more of the outstanding shares of Common Stock
or 50% or more of the voting power of the common equity of the Company, (iv) the Company, directly or indirectly, in one or more related
transactions effects any reclassification, reorganization or recapitalization of shares of Common Stock or any compulsory share exchange
pursuant to which shares of Common Stock are effectively converted into or exchanged for other securities, cash or property, or (v) the
Company, directly or indirectly, in one or more related transactions consummates a share purchase agreement or other business combination
(including, without limitation, a reorganization, recapitalization, spin-off or scheme of arrangement) with another Person or group of
Persons whereby such other Person or group acquires more than 50% of the outstanding shares of Common Stock or more of the outstanding
Common Stock or 50% or more of the voting power of the common equity of the Company (each a “Fundamental Transaction”),
then, upon any subsequent exercise of this Warrant, the Holder shall have the right to receive, for each Warrant Share that would have
been issuable upon such exercise immediately prior to the occurrence of such Fundamental Transaction, at the option of the Holder (without
regard to any limitation in Section 2(e) on the exercise of this Warrant), the number of shares of Common Stock of the successor or acquiring
corporation or of the Company, if it is the surviving corporation or is otherwise the continuing corporation, and any additional consideration
(the “Alternate Consideration”) receivable as a result of such Fundamental Transaction by a holder of the number of
shares of Common Stock for which this Warrant is exercisable immediately prior to such Fundamental Transaction (without regard to any
limitation in Section 2(e) on the exercise of this Warrant). For purposes of any such exercise, the determination of the Exercise Price
shall be appropriately adjusted to apply to such Alternate Consideration based on the amount of Alternate Consideration issuable in respect
of shares of Common Stock in such Fundamental Transaction, and the Company shall apportion the Exercise Price among the Alternate Consideration
in a reasonable manner reflecting the relative value of any different components of the Alternate Consideration. If holders of Common
Stock are given any choice as to the securities, cash or property to be received in a Fundamental Transaction, then the Holder shall
be given the same choice as to the Alternate Consideration it receives upon any exercise of this Warrant following such Fundamental Transaction.
The Company shall cause any successor entity in a Fundamental Transaction in which the Company is not the survivor (the “Successor
Entity”) to assume in writing all of the obligations of the Company under this Warrant in accordance with the provisions of
this Section 3(d) pursuant to written agreements prior to such Fundamental Transaction and shall, at the option of the Holder, deliver
to the Holder in exchange for this Warrant a security of the Successor Entity evidenced by a written instrument substantially similar
in form and substance to this Warrant which is exercisable for a corresponding number of shares or other securities of such Successor
Entity (or its parent entity) equivalent to the shares of Common Stock acquirable and receivable upon exercise of this Warrant (without
regard to any limitations on the exercise of this Warrant) prior to such Fundamental Transaction, and with an exercise price which applies
the exercise price hereunder to such shares of or other securities (but taking into account the relative value of the shares of Common
Stock pursuant to such Fundamental Transaction and the value of such shares or securities, such number of shares or securities and such
exercise price being for the purpose of protecting the economic value of this Warrant immediately prior to the consummation of such Fundamental
Transaction). Upon the occurrence of any such Fundamental Transaction, the Successor Entity shall succeed to, and be substituted for
(so that from and after the date of such Fundamental Transaction, the provisions of this Warrant referring to the “Company”
shall refer instead to the Successor Entity), and may exercise every right and power of the Company and shall assume all of the obligations
of the Company under this Warrant with the same effect as if such Successor Entity had been named as the Company herein.
Calculations.
All calculations under this Section 3 shall be made to the nearest cent or the nearest 1/100th of a share, as the case may be. For purposes
of this Section 3, the number of shares of Common Stock deemed to be issued and outstanding as of a given date shall be the sum of the
number of shares of Common Stock (excluding treasury shares, if any) issued and outstanding.
Notice
to Holder.
Adjustment
to Exercise Price. Whenever the Exercise Price is adjusted pursuant to any provision of this Section 3, the Company shall promptly
deliver to the Holder by facsimile or email a notice setting forth the Exercise Price after such adjustment and any resulting adjustment
to the number of Warrant Shares and setting forth a brief statement of the facts requiring such adjustment.
Notice
to Allow Exercise by Holder. If (A) the Company shall declare a dividend (or any other distribution in whatever form) on the Common
Stock, (B) the Company shall declare a special nonrecurring cash dividend on or a redemption of the Common Stock, (C) the Company shall
authorize the granting to all holders of the shares of Common Stock rights or warrants to subscribe for or purchase any shares of the
Company or of any rights, (D) the approval of any stockholders of the Company shall be required in connection with any reclassification
of the Common Stock, any consolidation or merger, amalgamation or arrangement to which the Company is a party, any sale or transfer of
all or substantially all of the assets of the Company, or any compulsory share exchange whereby the Common Stock is converted into other
securities, cash or property, or (E) the Company shall authorize the voluntary or involuntary dissolution, liquidation or winding up
of the affairs of the Company, then, in each case, the Company shall cause to be delivered by facsimile or email to the Holder at its
last facsimile number or email address as it shall appear upon the Warrant Register of the Company, at least 20 calendar days prior to
the applicable record or effective date hereinafter specified, a notice stating (x) the date on which a record is to be taken for the
purpose of such dividend, distribution, redemption, rights or warrants, or if a record is not to be taken, the date as of which the holders
of the Common Stock of record to be entitled to such dividend, distributions, redemption, rights or warrants are to be determined or
(y) the date on which such reclassification, consolidation, merger, amalgamation, arrangement, sale, transfer or share exchange is expected
to become effective or close, and the date as of which it is expected that holders of the Common Stock of record shall be entitled to
exchange their shares of Common Stock for securities, cash or other property deliverable upon such reclassification, consolidation, merger,
amalgamation, arrangement sale, transfer or share exchange; provided that the failure to deliver such notice or any defect therein or
in the delivery thereof shall not affect the validity of the corporate action required to be specified in such notice. To the extent
that any notice provided in this Warrant constitutes, or contains, material, non-public information regarding the Company or any of its
subsidiaries (the “Subsidiaries”), the Company shall simultaneously file such notice with the Commission pursuant
to a Current Report on Form 8-K. The Holder shall remain entitled to exercise this Warrant during the period commencing on the date of
such notice to the effective date of the event triggering such notice except as may otherwise be expressly set forth herein.
Transfer
of Warrant.
Transferability.
This Warrant and all rights hereunder are transferable, in whole or in part, upon surrender of this Warrant at the principal office of
the Company or its designated agent, together with a written assignment of this Warrant substantially in the form attached hereto duly
executed by the Holder or its agent or attorney and funds sufficient to pay any transfer taxes payable upon the making of such transfer.
Upon such surrender and, if required, such payment, the Company shall execute and deliver a new Warrant or Warrants in the name of the
assignee or assignees, as applicable, and in the denomination or denominations specified in such instrument of assignment, and shall
issue to the assignor a new Warrant evidencing the portion of this Warrant not so assigned, and this Warrant shall promptly be cancelled.
Notwithstanding anything herein to the contrary, the Holder shall not be required to physically surrender this Warrant to the Company
unless the Holder has assigned this Warrant in full, in which case, the Holder shall surrender this Warrant to the Company within three
(3) Trading Days of the date on which the Holder delivers an assignment form to the Company assigning this Warrant in full. This Warrant,
if properly assigned in accordance herewith, may be exercised by a new holder for the purchase of Warrant Shares without having a new
Warrant issued.
New
Warrants. This Warrant may be divided or combined with other Warrants upon presentation hereof at the aforesaid office of the Company,
together with a written notice specifying the names and denominations in which new Warrants are to be issued, signed by the Holder or
its agent or attorney. Subject to compliance with Section 4(a), as to any transfer which may be involved in such division or combination,
the Company shall execute and deliver a new Warrant or Warrants in exchange for the Warrant or Warrants to be divided or combined in
accordance with such notice. All Warrants issued on transfers or exchanges shall be dated the original Issue Date of this Warrant and
shall be identical with this Warrant except as to the number of Warrant Shares issuable pursuant thereto.
Warrant
Register. The Company shall register this Warrant, upon records to be maintained by the Company for that purpose (the “Warrant
Register”), in the name of the record Holder hereof from time to time. The Company may deem and treat the registered Holder
of this Warrant as the absolute owner hereof for the purpose of any exercise hereof or any distribution to the Holder, and for all other
purposes, absent actual notice to the contrary.
Miscellaneous.
No
Rights as Stockholder Until Exercise. This Warrant does not entitle the Holder to any voting rights, dividends or other rights as
a stockholder of the Company prior to the exercise hereof as set forth in Section 2(d)(i), except as expressly set forth in Section 3.
Without limiting any rights of a Holder to receive Warrant Shares on a “cashless exercise” pursuant to Section 2(c) or to
receive cash payments pursuant to Section 2(d)(i) and Section 2(d)(iv) herein, in no event, including if the Company is for any reason
unable to issue and deliver Warrant Shares upon exercise of this Warrant as required pursuant to the terms thereof, shall the Company
be required to net cash settle an exercise of this Warrant.
Loss,
Theft, Destruction or Mutilation of Warrant. The Company covenants that upon receipt by the Company of evidence reasonably satisfactory
to it of the loss, theft, destruction or mutilation of this Warrant or any stock certificate relating to the Warrant Shares, and in case
of loss, theft or destruction, of indemnity or security reasonably satisfactory to it (which shall in no event include the posting of
any bond), and upon surrender and cancellation of such Warrant or stock certificate, if mutilated, the Company shall make and deliver
a new Warrant or stock certificate of like tenor and dated as of such cancellation, in lieu of such Warrant or stock certificate.
Saturdays,
Sundays, Holidays, etc. If the last or appointed day for the taking of any action or the expiration of any right required or granted
herein shall not be a Trading Day, then, such action may be taken or such right may be exercised on the next succeeding Trading Day.
Authorized
Shares.
The
Company covenants that, during the period the Warrant is outstanding, it will reserve from its authorized and unissued shares of Common
Stock a sufficient number of shares to provide for the issuance of the Warrant Shares upon the exercise of any purchase rights under
this Warrant. The Company further covenants that its issuance of this Warrant shall constitute full authority to its officers who are
charged with the duty of issuing the necessary Warrant Shares upon the exercise of the purchase rights under this Warrant. The Company
shall take all such reasonable action as may be necessary to assure that such Warrant Shares may be issued as provided herein without
violation of any applicable law or regulation, or of any requirements of the Trading Market upon which the Common Stock may be listed
or quoted for trading. The Company covenants that all Warrant Shares which may be issued upon the exercise of the purchase rights represented
by this Warrant will, upon exercise of the purchase rights represented by this Warrant and payment for such Warrant Shares in accordance
herewith, be duly authorized, validly issued, fully paid and non-assessable and free from all taxes, liens and charges created by the
Company in respect of the issue thereof (other than taxes in respect of any transfer occurring contemporaneously with such issue).
Except
and to the extent as waived or consented to by the Holder, the Company shall not by any action, including, without limitation, amending
its certificate of incorporation or through any reorganization, transfer of assets, consolidation, merger, amalgamation, arrangement
dissolution, issue or sale of securities or any other voluntary action, avoid or seek to avoid the observance or performance of any of
the terms of this Warrant, but will at all times in good faith assist in the carrying out of all such terms and in the taking of all
such actions as may be necessary or appropriate to protect the rights of Holder as set forth in this Warrant against impairment. Without
limiting the generality of the foregoing, the Company shall (i) not increase the par value of any Warrant Shares above the amount payable
therefor upon such exercise immediately prior to such increase in par value, (ii) take all such action as may be necessary or appropriate
in order that the Company may validly and legally issue fully paid and non-assessable Warrant Shares upon the exercise of this Warrant
and (iii) use commercially reasonable efforts to obtain all such authorizations, exemptions or consents from any public regulatory body
having jurisdiction thereof, as may be, necessary to enable the Company to perform its obligations under this Warrant.
Before
taking any action which would result in an adjustment in the number of Warrant Shares for which this Warrant is exercisable or in the
Exercise Price, the Company shall obtain all such authorizations or exemptions thereof, or consents thereto, as may be necessary from
any public regulatory body or bodies having jurisdiction thereof.
Governing
Law. All questions concerning the construction, validity, enforcement and interpretation of this Warrant shall be governed by and
construed and enforced in accordance with the internal laws of the State of New York, without regard to the principles of conflict of
laws thereof. Each party agrees that all legal Proceedings concerning the interpretation, enforcement and defense of this Warrant shall
be commenced in the state and federal courts sitting in the City of New York, Borough of Manhattan (the “New York Courts”).
Each party hereto hereby irrevocably submits to the exclusive jurisdiction of the New York Courts for the adjudication of any dispute
hereunder or in connection herewith or with any transaction contemplated hereby or discussed herein (including with respect to the enforcement
of any provision hereunder), and hereby irrevocably waives, and agrees not to assert in any suit, action or Proceeding, any claim that
it is not personally subject to the jurisdiction of such New York Courts, or such New York Courts are improper or inconvenient venue
for such Proceeding. If any party shall commence an action or Proceeding to enforce any provisions of this Warrant, then the prevailing
party in such action or Proceeding shall be reimbursed by the other party for its attorneys’ fees and other costs and expenses
incurred in the investigation, preparation and prosecution of such action or Proceeding.
Restrictions.
The Holder acknowledges that the Warrant Shares acquired upon the exercise of this Warrant, if not registered, and the Holder does not
utilize cashless exercise, will have restrictions upon resale imposed by state and federal securities laws.
Nonwaiver
and Expenses. No course of dealing or any delay or failure to exercise any right hereunder on the part of Holder shall operate as
a waiver of such right or otherwise prejudice the Holder’s rights, powers or remedies. Without limiting any other provision of
this Warrant, if the Company willfully and knowingly fails to comply with any provision of this Warrant, which results in any material
damages to the Holder, the Company shall pay to the Holder such amounts as shall be sufficient to cover any costs and expenses including,
but not limited to, reasonable attorneys’ fees, including those of appellate Proceedings, incurred by the Holder in collecting
any amounts due pursuant hereto or in otherwise enforcing any of its rights, powers or remedies hereunder.
Notices.
Any and all notices or other communications or deliveries to be provided by the Holders hereunder including, without limitation, any
Notice of Exercise, shall be in writing and delivered personally, by facsimile or by e-mail, or sent by a nationally recognized overnight
courier service, addressed to the Company, 21 Business Park Drive, Branford, CT 06405, Attention: Chief Financial Officer, email address:
__________ or such other email address or address as the Company may specify for such purposes by notice to the Holders. Any and all
notices or other communications or deliveries to be provided by the Company hereunder shall be in writing and delivered personally, by
facsimile, email or sent by a nationally recognized overnight courier service addressed to each Holder at the facsimile number, email
address or address of such Holder appearing on the books of the Company. Any notice or other communication or deliveries hereunder shall
be deemed given and effective on the earliest of (i) the time of transmission, if such notice or communication is delivered via facsimile
at the facsimile number or e-mail at the e-mail address set forth in this Section prior to 5:30 p.m. (New York City time) on any date,
(ii) the next Trading Day after the date of transmission, if such notice or communication is delivered via facsimile at the facsimile
number or e-mail at the e-mail address set forth in this Section on a day that is not a Trading Day or later than 5:30 p.m. (New York
City time) on any Trading Day, (iii) the second Trading Day following the date of mailing, if sent by U.S. nationally recognized overnight
courier service, or (iv) upon actual receipt by the party to whom such notice is required to be given.
Limitation
of Liability. No provision hereof, in the absence of any affirmative action by the Holder to exercise this Warrant to purchase Warrant
Shares, and no enumeration herein of the rights or privileges of the Holder, shall give rise to any liability of the Holder for the purchase
price of any Common Stock or as a stockholder of the Company, whether such liability is asserted by the Company or by creditors of the
Company.
Remedies.
The Holder, in addition to being entitled to exercise all rights granted by law, including recovery of damages, will be entitled to specific
performance of its rights under this Warrant. The Company agrees that monetary damages would not be adequate compensation for any loss
incurred by reason of a breach by it of the provisions of this Warrant and hereby agrees to waive and not to assert the defense in any
action for specific performance that a remedy at law would be adequate.
Successors
and Assigns. Subject to applicable securities laws, this Warrant and the rights and obligations evidenced hereby shall inure to the
benefit of and be binding upon the successors and permitted assigns of the Company and the successors and permitted assigns of Holder.
The provisions of this Warrant are intended to be for the benefit of any Holder from time to time of this Warrant and shall be enforceable
by the Holder or holder of Warrant Shares.
Amendment.
This Warrant may be modified or amended or the provisions hereof waived with the written consent of the Company, on the one hand, and
the Holder on the other hand.
Severability.
Wherever possible, each provision of this Warrant shall be interpreted in such manner as to be effective and valid under applicable law,
but if any provision of this Warrant shall be prohibited by or invalid under applicable law, such provision shall be ineffective to the
extent of such prohibition or invalidity, without invalidating the remainder of such provisions or the remaining provisions of this Warrant.
No
Expense Reimbursement. The Holder shall in no way be required the pay, or to reimburse the Company for, any fees or expenses of the
Company’s transfer agent in connection with the issuance or holding or sale of the Common Stock, Warrant and/or Warrant Shares.
The Company shall solely be responsible for any and all such fees and expenses.
Headings.
The headings used in this Warrant are for the convenience of reference only and shall not, for any purpose, be deemed a part of this
Warrant.
********************
(Signature
Page Follows)
IN
WITNESS WHEREOF, the Company has caused this Warrant to be executed by its officer thereunto duly authorized as of the date first above
indicated.
| |
Azitra,
Inc. |
| |
|
|
| |
By:
|
|
| |
Name: |
|
| |
Title: |
|
NOTICE
OF EXERCISE
(1)
The undersigned hereby elects to purchase ________ Warrant Shares of the Company pursuant to the terms of the attached Warrant (only
if exercised in full), and tenders herewith payment of the exercise price in full, together with all applicable transfer taxes, if any.
(2)
Payment shall take the form of (check applicable box):
☐
in lawful money of the United States; or
☐
the cancellation of such number of Warrant Shares as is necessary, in accordance with the formula set forth in subsection 2(c), to exercise
this Warrant with respect to the maximum number of Warrant Shares purchasable pursuant to the cashless exercise procedure set forth in
subsection 2(c).
(3)
Please issue said Warrant Shares in the name of the undersigned or in such other name as is specified below:
The
Warrant Shares shall be delivered to the following DWAC Account Number:
[SIGNATURE
OF HOLDER]
| Name
of Investing Entity: |
|
|
| |
|
|
| Signature
of Authorized Signatory of Investing Entity: |
|
|
| |
|
|
| Name
of Authorized Signatory: |
|
|
| |
|
|
| Title
of Authorized Signatory: |
|
|
ASSIGNMENT
FORM
(To
assign the foregoing Warrant, execute this form and supply required information. Do not use this form to purchase shares.)
FOR
VALUE RECEIVED, the foregoing Warrant and all rights evidenced thereby are hereby assigned to
| Name: |
|
| |
(Please
Print) |
| |
|
| Address: |
|
| |
(Please
Print) |
| |
|
| Phone
Number: |
|
| |
|
| Email
Address: |
|
Dated:
_____________________ __, ______
| Holder’s
Signature: |
|
|
| |
|
|
| Holder’s
Address: |
|
|
Exhibit 10.4
EMPLOYMENT
AGREEMENT
EMPLOYMENT
AGREEMENT effective as of July 5, 2023 (this “Agreement”) between Azitra Inc. (the “Company”),
a Delaware corporation, and Travis Whitfill (the “Executive”) (the Company and the Executive collectively, the “Parties”).
Background:
The
Parties desire to enter into this Agreement to provide for the employment of the Executive by the Company and to govern the terms and
conditions of the Executive’s employment by the Company.
Terms:
NOW,
THEREFORE, in consideration of the premises and covenants set forth herein, and intending to be legally bound hereby, the Parties hereby
agree as follows:
1. Position and Duties.
(a) Position
and Duties. The Company agrees that the Executive shall be employed by the Company to serve as Chief Operating Officer of the Company.
The Executive shall report to the President and Chief Executive Officer of the Company (the “CEO”). The Executive
agrees to be so employed by the Company and agrees to devote substantially all of his business time, attention, skill and efforts to
perform services for the Company and to faithfully and diligently discharge and fulfill his duties hereunder to the best of his abilities.
In so doing, the Executive shall perform such executive, managerial, administrative and financial functions as are required to develop
the Company’s business and to perform other duties assigned to the Executive by the CEO and/or the Board of Directors of the Company
(the “Board”). Executive will perform services under this Agreement remotely, with the understanding that he will
appear in the Company offices and travel for business as may reasonably be necessary.
(b) Other
Activities. Notwithstanding Section 1(a), nothing contained in Agreement shall prevent or limit: (i) the Executive’s
right to manage the Executive’s personal investments or personal financial or legal matters including, without limitation, the
right to make passive investments in the securities of (A) any entity which the Executive does not control, directly or indirectly, and
which does not compete with Company, or (B) any publicly held entity, so long as the Executive’s aggregate direct and indirect
interest does not exceed two percent (2%) of the issued and outstanding securities of any class of securities of such publicly held entity;
(ii) subject to prior approval of the CEO and the Board, the Executive’s right to serve as a member of the board of directors of
up to two (2) entities that do not compete with the Company; or (iii) the Executive’s participation in civic, nonprofit, political
and charitable activities, including, subject to prior approval of the CEO and the Board, which shall not be unreasonably withheld, as
a member of a board of a civic, political or charitable organization.
2. Term.
The Executive’s employment under this Agreement shall commence on June 21, 2023 (the “Effective Date”) and shall
continue until terminated pursuant to Section 4 (the “Term”).
3.
Compensation.
(a) Base
Salary. During the Term, the Executive shall be paid a base salary at the annual rate of Three Hundred Fifty Thousand Dollars ($350,000.00)
(the “Base Salary”), payable in accordance with the Company’s payroll practices and policies in effect from
time to time and subject to applicable withholding of income taxes, social security taxes and other such other payroll deductions as
are required by law or applicable employee benefit programs. The Base Salary will be subject to periodic review and increase at the sole
discretion of the CEO and the Board.
(b) Annual
Bonus. With respect to each fiscal year of the Company completed during the Term, the Executive will be eligible to be considered
for an annual performance bonus (the “Annual Bonus”) in an amount of up to 30% of the Executive’s Base Salary;
provided, however, that any Annual Bonus for the 2023 fiscal year shall be prorated based on the portion of such fiscal year during which
the Executive is actually employed by the Company pursuant to this Agreement. The Annual Bonus, if any, will be awarded by the CEO and
Board in their sole discretion based on the achievement of Company and personal performance metrics established by the CEO and Board
on an annual basis, and in any event not later than the 90th day of the applicable fiscal year. Any Annual Bonus allocable to the Executive
hereunder shall be earned by the Executive if and only if the Executive is actively employed on a full-time basis with the Company and
is otherwise in compliance with the Executive’s obligations under this Agreement through the end of the fiscal year to which such
Annual Bonus relates and, subject to Section 4 hereof, on the payment date therefor. Any Annual Bonus awarded to the Executive
hereunder will be payable in a single lump sum cash payment, less applicable taxes and withholdings, not later than three and one-half
months after the end of the fiscal year to which it relates in accordance with the Company’s customary practices for annual bonus
payments.
(c) Vacation
and Fringe Benefits. During the Term, the Executive shall be eligible for four (4) weeks of paid vacation per calendar year, accruing
in accordance with the vacation policies established by the Company from time to time. During the Term, the Executive also shall be entitled
to participate in all of the Company’s then-existing employee benefit programs for which senior management employees of the Company
are generally eligible. Nothing in this Agreement will preclude the Company from changing, altering or terminating any of the plans or
programs for which employees of the Company are eligible, in whole or in part, in the Company’s sole discretion.
(d) Equity
Opportunity. The Company agrees to discuss in good faith with Executive a potential future grant of equity in the Company prior to
the end of 2023 or, if the Company has not adopted a new equity plan by the end of 2023, shortly following the Company’s adoption
of a new equity plan.
(e) Reimbursement
of Expenses. During the Term, and subject to Section 16(c) hereunder, the Company shall reimburse the Executive for the reasonable
and necessary out-of-pocket travel and other business expenses incurred by the Executive for or on behalf of the Company in furtherance
of the performance of the Executive’s duties hereunder in accordance with the Company’s reimbursement policies as approved
by the Board from time to time, subject in all cases to the Company’s requirements with respect to reporting and documentation
of such expenses.
4.
Termination.
(a) Death.
This Agreement and Executive’s employment with the Company shall automatically terminate effective as of the date of the Executive’s
death, in which event the Company shall not have any further obligation or liability under this Agreement except that the Company shall
pay to the Executive’s estate: (i) any portion of the Executive’s Base Salary for the period up to the Executive’s
date of death that has been earned but remains unpaid; (ii) any unpaid expense reimbursement owed to the Executive under Section 3(e);
(iii) any amount earned, accrued and arising from the Executive’s participation in, or benefits accrued under, any Company employee
benefit plan or arrangement, which amounts shall be payable in accordance with the terms and conditions of such employee benefit plans
and arrangements (the payments in clauses (i) through (iii) collectively, the “Accrued Obligations”); and (iv) the
Annual Bonus awarded pursuant to Section 3(b), if any, with respect to the fiscal year prior to the fiscal year of termination,
to the extend unpaid. The Annual Bonus, if any, will be paid when it would have been paid had the Executive remained employed with the
Company.
(b) Incapacity.
The Company may terminate this Agreement and the employment of the Executive immediately upon written notice to the Executive in the
event of the Incapacity of the Executive, in which event the Company shall not have any further obligation or liability under this Agreement
except for the Accrued Obligations and the Annual Bonus awarded pursuant to Section 3(b), if any, with respect to the fiscal year
prior to the fiscal year of termination, to the extend unpaid. The Annual Bonus, if any, will be paid when it would have been paid had
the Executive remained employed with the Company. For purposes of this Agreement, “Incapacity” shall mean the Executive’s
incapacity or inability to perform the Executive’s duties and responsibilities as contemplated herein for one hundred twenty (120)
days or more within any one (1) year period (cumulative or consecutive), because the Executive’s physical or mental health has
become so impaired as to make it impossible or impractical for the Executive to perform the duties and responsibilities contemplated
hereunder with or without accommodation. The Executive’s physical or mental health shall be determined by a medical expert appointed
by mutual agreement between the Company and the Executive following such expert’s examination of the Executive. The Executive hereby
consents to such examination and consultation regarding the Executive’s health and ability to perform as aforesaid.
(c) Termination
of the Executive’s Employment for Cause. The Company may terminate this Agreement and the employment of the Executive for Cause
immediately upon providing written notice of such termination to the Executive. If the Executive’s employment with the Company
is terminated by the Company for Cause, the Company shall not have any further obligation or liability under this Agreement except for
the Accrued Obligations. For purposes of this Agreement, “Cause” shall mean with respect to the Executive one or more
of the following: (i) the Executive’s commission at any time of any act or omission that results in, or that may reasonably be
expected to result in, a conviction, plea of no contest or imposition of unadjudicated probation for any felony or for any crime involving
moral turpitude; (ii) severe, continued or habitual intoxication, or reporting to work under the influence of alcohol or illegal drugs;
(iii) conduct that causes, or could reasonably be expected to cause, the Company reputational or economic harm; (iv) the Executive’s
failure to satisfactorily perform the duties of the Executive’s position, provided that Executive fails to cure such failure to
perform, to the satisfaction of the Board in its sole discretion, within thirty (30) days following notice from the Company thereof;
(v) any act or omission aiding or abetting a competitor, supplier or customer of the Company and/or any of its subsidiaries or affiliates
to the disadvantage or detriment of the Company and/or any of its subsidiaries or affiliates; (vi) breach of fiduciary duty or duty of
loyalty; (vii) the Executive’s failure to observe and comply with any of the Company’s rules, policies and/or procedures,
including, but not limited to, the Company’s policies prohibiting discrimination, harassment or retaliation, in the Company’s
sole discretion; (viii) willful misconduct, fraud, misappropriation, embezzlement, gross negligence, self-dealing, dishonesty or misrepresentation;
(ix) the Executive’s failure to follow the lawful instructions of the CEO or the Board; or (x) any material breach of this Agreement
or any other agreement between the Company and the Executive (including, but not limited to, the Covenants Agreement (as defined below).
(d) Other
Termination by the Company. The Company may terminate this Agreement and the employment of the Executive for any reason other than
Incapacity or Cause immediately upon written notice of termination to the Executive. If the Executive’s employment with the Company
is terminated by the Company for any reason other than Incapacity or Cause within the six (6) month period following the Effective Date,
the Company shall not have any further obligation or liability under this Agreement except for the Accrued Obligations. If the Executive’s
employment with the Company is terminated by the Company for any reason other than Incapacity or Cause more than six (6) months following
the Effective Date, then in addition to the Accrued Obligations, and subject to the execution by the Executive (and the irrevocability)
of a release of claims in a form satisfactory to the Company (the “Release”), the compliance by the Executive with
the Release, the Covenants Agreement (as defined below), and the compliance by the Executive with all terms and provisions of this Agreement
that survive the termination of the Executive’s employment by the Company, the Executive shall be entitled to receive: (i) an amount
equal to the Base Salary (as in effect immediately prior to termination of employment) for a period of six (6) months following the date
of termination of employment, paid in equal installments over a six (6) month period in accordance with the Company’s payroll schedule
in effect at the time (the “Severance Payments”); and (ii) the Annual Bonus awarded pursuant to Section 3(b),
if any, with respect to the fiscal year prior to the fiscal year of termination, to the extend unpaid, when it would have been paid had
the Executive remained employed with the Company. The Executive shall not be entitled to any other salary, compensation or other benefits
after termination of the Executive’s employment, except as specifically provided for in the Company’s employee benefit plans
or as otherwise expressly required by applicable law.
Any
Severance Payments payable pursuant to this Section 4(d) shall not be paid until the first scheduled payment date following the
date the Release is executed and no longer subject to revocation, with the first such payment being in an amount equal to the total amount
to which the Executive would otherwise have been entitled during the period following the date of termination if such deferral had not
been required; provided, however, that any such amounts that constitute nonqualified deferred compensation within the meaning
of Internal Revenue Code Section 409A and the regulations and guidance promulgated thereunder (“Section 409A”) shall
not be paid until the 60th day following such termination to the extent necessary to avoid adverse tax consequences under Section 409A
(provided that, if the 60-day period begins in one taxable year and ends in a second taxable year, such Severance Payments shall not
commence until the second taxable year), and, if such payments are required to be so deferred, the first payment shall be in an amount
equal to the total amount to which the Executive would otherwise have been entitled during the period following the date of termination
if such deferral had not been required; provided further that, if the Executive is a “specified employee” within the
meaning of Section 409A, any Severance Payments payable to the Executive under this Section 4(d) during the first six months and
one day following the date of termination pursuant to this Section 4(d) that constitute nonqualified deferred compensation within
the meaning of Section 409A shall not be paid until the date that is six (6) months and one day following such termination to the extent
necessary to avoid adverse tax consequences under Section 409A, and, if such payments are required to be so deferred, the first payment
shall be in an amount equal to the total amount to which the Executive would otherwise have been entitled to during the period following
the date of termination if such deferral had not been required.
(e) Termination
by the Executive. The Executive may terminate this Agreement and the Executive’s employment with the Company for any reason
upon thirty (30) days’ prior written notice of termination to the Company. In the event the Executive terminates the Executive’s
employment hereunder, the Company shall not have any further obligation or liability under this Agreement, except for the Accrued Obligations,
which shall be paid on the first payroll date following last date of employment to the extent administratively feasible and if not, then
on the second payroll date following the last date of employment (unless otherwise required by applicable law). If the Executive terminates
his employment with the Company, the Company, in its sole discretion, may terminate the Executive’s employment prior to the effective
date of the Executive’s resignation, provided that the Company pays the Executive his Base Salary for the thirty (30) day period
following the notice of termination.
(f) No
Other Payments or Benefits Owing. The payments and benefits set forth in this Section 4 shall be the sole amounts owing to
the Executive upon termination of the Executive’s employment for the reasons set forth above and the Executive shall not be eligible
for any other payments or other forms of compensation or benefits. The Company may offset any amounts the Executive owes the Company
against any amounts the Company owes the Executive hereunder.
(g) If
the Executive breaches any provision of this Agreement or the Covenants Agreement, the Company (i) shall no longer be obligated to make
any payments or provide any other benefits pursuant to this Section 4, and (ii) shall be entitled to reimbursement of any Severance
Payments made to the Executive pursuant to Section 4(d).
5. Non-Competition,
Non-Solicitation, Non-Disclosure Agreement. In light of the competitive and proprietary aspects of the business of Company, and as
a condition of Executive’s employment hereunder, Executive agrees to sign and abide by Company’s Non- Competition, Non-Solicitation,
Non-Disclosure, and Inventions Assignment Agreement (the “Covenants Agreement”).
6. No
Conflicts. The Executive represents and warrants that the Executive is not party to any agreement, contract or understanding, whether
of employment, consultancy or otherwise, in conflict with this Agreement or which would in any way restrict or prohibit the Executive
from undertaking or performing services for the Company or otherwise from entering into or performing this Agreement or the Covenants
Agreement.
7. Full
Agreement. This Agreement (and the Covenants Agreement) constitutes the entire agreement of the Parties concerning its subject matter
and supersedes all other oral or written understandings, discussions, and agreements, and may be modified only in a writing signed by
both Parties. The Parties acknowledge that they have read and fully understand the contents of this Agreement and execute it after having
an opportunity to consult with legal counsel.
8. Amendment
and Waiver. The terms and provisions of this Agreement may be modified or amended only by written agreement executed by the Parties
hereto. The terms and provisions of this Agreement may be waived, or consent for the departure therefrom granted, only by written document
executed by the party entitled to the benefits of such terms or provisions. No such waiver or consent shall be deemed to be or shall
constitute a waiver or consent with respect to any other terms or provisions of this Agreement, whether or not similar. Each such waiver
or consent shall be effective only in the specific instance and for the purpose for which it was given, and shall not constitute a continuing
waiver or consent.
9. Enforceability.
If any provision of this Agreement shall be invalid or unenforceable, in whole or in part, then such provision shall be deemed to be
modified or restricted to the extent and in the manner necessary to render the same valid and enforceable, or shall be deemed excised
from this Agreement, as the case may require, and this Agreement shall be construed and enforced to the maximum extent permitted by law
as if such provision had been originally incorporated herein as so modified or restricted or as if such provision had not been originally
incorporated herein, as the case may be.
10. No
Strict Construction. The language used in this Agreement shall be deemed to be the language chosen by the parties hereto to express
their mutual intent, and no rule of strict construction shall be applied against any party.
11. Governing
Law and Venue. This Agreement will be construed under the laws of the State of Connecticut, without regard to its conflict of law
provisions, and the parties irrevocably consent and agree that the federal and state courts of New Haven County, Connecticut will have
exclusive jurisdiction over any dispute relating to the Executive’s employment with the Company or to this Agreement.
12.
Assignment.
(a) By
the Company. The rights and obligations of the Company under this Agreement shall inure to the benefit of, and shall be binding upon,
the successors and assigns of the Company. This Agreement and any of the rights or obligations hereunder may be assigned by the Company
without the consent of the Executive including, but not limited to, in connection with the sale, merger, consolidation, reorganization,
liquidation or transfer, in whole or in part, of the Company’s control and/or ownership of its assets or business. In such event,
the Executive agrees to continue to be bound by the terms of this Agreement.
(b) By
the Executive. This Agreement and the obligations created hereunder may not be assigned by the Executive, but all rights of the Executive
hereunder shall inure to the benefit of and be enforceable by the Executive’s heirs, devisees, legatees, executors, administrators
and personal representatives. Any attempted assignment in violation of this Section 12(b) shall be null and void.
13. Notices.
Except as otherwise specifically provided herein, any notice required or permitted by this Agreement shall be in writing and shall be
delivered as follows with notice deemed given as indicated: (a) by personal delivery when delivered personally; (b) by overnight courier
upon written verification of receipt; (c) by facsimile transmission upon acknowledgment of receipt of electronic transmission; or (d)
by certified or registered mail, return receipt requested, upon verification of receipt.
If
to the Company:
Azitra
Inc.
21
Business Park Drive
Branford, CT 06405
Attention:
President and Chief Executive Officer
Attention: Chair, Board of Directors
With
a copy to:
Duane
Morris LLP
30 S. 17th Street
Philadelphia,
PA 19103
Attention: Kathleen M. Shay, Esq.
If
to the Executive:
Travis
Whitfill
215 E. 68th Street
Apt. 33J
New
York, NY 10065
Any
party may from time to time change its address for the purpose of notices to that party by a similar notice specifying a new address,
but no such change shall be deemed to have been given until it is actually received by the party sought to be charged with its contents.
14. Executive’s
Cooperation. During the Term and thereafter, the Executive shall reasonably cooperate with the Company in any internal investigation
or administrative, regulatory or judicial proceeding as reasonably requested by the Company (including, without limitation, the Executive
being reasonably available to the Company upon reasonable notice for interviews and factual investigations, appearing at the Company’s
reasonable request to give testimony without requiring service of a subpoena or other legal process, volunteering to the Company all
pertinent information and turning over to the Company all relevant documents which are or may come into the Executive’s possession)
at reasonable times. In the event the Company requires the Executive’s cooperation in accordance with this Section 14, the
Company shall reimburse the Executive solely for reasonable travel expenses incurred by the Executive as a result of such cooperation
(including lodging and meals, upon submission of receipts).
Nothing
about the foregoing shall preclude the Executive from testifying truthfully in any forum or from providing truthful information to any
government agency or commission.
15. Counterparts.
This Agreement may be executed in separate counterparts (including by means of telecopied signature pages or electronic transmission
in portable document format (pdf)), each of which is deemed to be an original and all of which taken together constitute one and the
same agreement.
16.
409A Compliance.
(a) The
intent of the Parties is that payments and benefits under this Agreement comply with Section 409A and, accordingly, to the maximum extent
permitted, this Agreement shall be interpreted to be in compliance therewith. To the extent such potential payments or benefits could
become subject to such additional taxes under Section 409A, the Parties shall cooperate to attempt to amend this Agreement with the goal
of giving the Executive the economic benefits described herein in a manner that does not result in such tax being imposed. In no event
shall the Company or its subsidiaries or affiliates be liable for any additional tax, interest or penalty that may be imposed on the
Executive under Section 409A or damages for failing to comply with Section 409A.
(b) A
termination of employment shall not be deemed to have occurred for purposes of any provision of this Agreement providing for the payment
of any amounts or benefits upon or following a termination of employment unless such termination is also a “separation from service”
within the meaning of Section 409A and, for purposes of any such provision of this Agreement, references to a “termination,”
“termination of employment” or like terms shall mean “separation from service.”
(c) To
the extent that reimbursements or other in-kind benefits under this Agreement constitute “nonqualified deferred
compensation” for purposes of Section 409A, (i) all such expenses or other reimbursements hereunder shall be made on or prior
to the last day of the taxable year following the taxable year in which such expenses were incurred by the Executive, (ii) any such
right to reimbursement or in-kind benefits shall not be subject to liquidation or exchange for another benefit, and (iii) no such
reimbursement, expenses eligible for reimbursement, or in-kind benefits provided in any taxable year shall in any way affect the
expenses eligible for reimbursement, or in-kind benefits to be provided, in any other taxable year.
(d) For
purposes of Section 409A, the Executive’s right to receive any installment payments pursuant to this Agreement shall be treated
as a right to receive a series of separate and distinct payments and each payment shall be treated as a separate payment.
(e) Notwithstanding
any other provision of this Agreement to the contrary, in no event shall any payment under this Agreement that constitutes “nonqualified
deferred compensation” for purposes of Section 409A be subject to offset by any other amount unless otherwise permitted by Section
409A.
17.
Survival. The provisions of Section 4 through this Section 17, inclusive, shall survive and continue in full force
in accordance with their terms notwithstanding the termination of the Term or the Executive’s employment with the
Company.
(Signature
page follows.)
IN
WITNESS WHEREOF, this Agreement has been executed by the Parties as of the date first above written.
| |
AZITRA INC. |
| |
|
| |
By: |
 |
| |
Name: |
Francisco
Salva |
| |
Title: |
President
and Chief Executive Officer |
| |
 |
| |
Travis
Whitfill |
Exhibit
23.2

Consent
of Independent Registered Public Accounting Firm
We
hereby consent to the inclusion in this Registration Statement on Form S-1 of our report dated February 20, 2023, except for Notes 9,
10, 11, 14, and 19, as to which the date is June 13, 2023, which includes an explanatory paragraph as to the Company’s ability
to continue as a going concern, relating to the financial statements of Azitra, Inc. as of and for the years ended December 31, 2022
and 2021. We also consent to the reference to our firm under the heading “Experts” appearing therein.
/s/
Grassi & Co., CPAs, P.C.
Grassi
& Co., CPAs, P.C.
Jericho,
New York
January
19, 2024
Exhibit
107
Calculation
of Filing Fee Tables
Form
S-1
(Form Type)
Azitra
Inc.
(Exact Name of Registrant as Specified in its Charter)
Table
1: Newly Registered and Carry Forward Securities
| | |
Security
Type | |
Security
Class
Title | |
Fee
Calculation
or Carry
Forward
Rule | | |
Amount
Registered (a) | | |
Proposed
Maximum
Offering
Price Per Unit | | |
Maximum
Aggregate
Offering
Price (b) | | |
Fee Rate | | |
Amount of
Registration
Fee | |
Fees to Be
Paid | |
Equity | |
Common stock | |
| 457 (o) | | |
| | | |
| | | |
$ | 7,072,500 | | |
| $147.60 per $1,000,000 | | |
$ | 1,043.91 | |
| | |
Equity | |
Warrants to be issued to the representative of the underwriters (c) | |
| (d) | | |
| | | |
| | | |
| | | |
| | | |
| | |
| | |
Equity | |
Pre-Funded Warrants | |
| (e) | | |
| | | |
| | | |
| | | |
| | | |
| | |
| | |
Equity | |
Common stock underlying warrants to be issued to the representative of the underwriters | |
| 457 (o) | | |
| | | |
| | | |
$ | 353,625 | | |
| $147.60 per $1,000,000 | | |
$ | 52.20 | |
| | |
Equity | |
Common stock underlying Pre-Funded Warrants | |
| (e) | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | |
Fees
Previously
Paid | |
| |
— | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | |
| | |
| |
| |
| | | |
| | | |
| | | |
| | | |
| | | |
| | |
| | |
Total Offering Amounts | |
| |
| | | |
| | | |
| | | |
$ | 7,426,125 | | |
| | | |
$ | 1,096.11 | |
| | |
Total Fees Previously Paid | |
| |
| | | |
| | | |
| | | |
| | | |
| | | |
$ | -0- | |
| | |
Total Fee Offsets | |
| |
| | | |
| | | |
| | | |
| | | |
| | | |
| - | |
| | |
Net Fee Due | |
| |
| | | |
| | | |
| | | |
| | | |
| | | |
$ | 1,096.11 | |
(a) In accordance with Rule 416 under the Securities Act of 1933, as amended (the “Securities Act”), this registration statement
shall be deemed to cover an indeterminate number of additional shares of Common Stock to be offered or issued from stock splits, stock
dividends or similar transactions.
(b)
The proposed maximum aggregate offering price has been estimated solely to calculate the registration fee in accordance with Rule 457(o)
under the Securities Act.
(c)
We have agreed to issue to the representative of the underwriters warrants to purchase shares of common stock representing up to 4% of
the common stock issued in the offering. The representative’s warrants are exercisable at a per share exercise price equal to 125%
of the public offering price per share of the common stock offered hereby.
(d)
No registration fee required pursuant to Rule 457(g).
(e)
The registrant may issue pre-funded warrants to purchase shares of common stock in the offering. The purchase price of each pre-funded
warrant will equal the price per share at which shares of common shares are being sold to the public in this offering, minus $0.001,
which constitutes the pre-funded portion of the exercise price, and the remaining unpaid exercise price of the pre-funded warrant
will equal $0.001 per share (subject to adjustment as provided for therein). The proposed maximum aggregate offering price of
the common stock will be reduced on a dollar-for-dollar basis based on the offering price of any pre-funded warrants issued in the offering,
and the proposed maximum aggregate offering price of the pre-funded warrants to be issued in the offering will be reduced on a dollar-for-dollar
basis based on the offering price of any common stock issued in the offering. Accordingly, the proposed maximum aggregate offering price
of the common stock and pre-funded warrants (including the common stock issuable upon exercise of the pre-funded warrants), if any, is
$7,072,500. Includes the offering price of shares of common stock and/or pre-funded warrants that the representative of the underwriter
has the option to purchase to cover over- allotments, if any.
Grafico Azioni Azitra (AMEX:AZTR)
Storico
Da Ott 2024 a Nov 2024

Grafico Azioni Azitra (AMEX:AZTR)
Storico
Da Nov 2023 a Nov 2024
